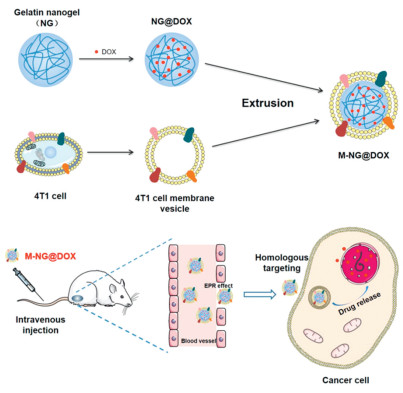
Scheme 1.
Preparation of 4T1 cell membrane camouflaged biomimetic nanoplatform M-NG@DOX with homologous targeting ability for cancer therapy.
Cancer cell membrane camouflaged biomimetic gelatin-based nanogel for tumor inhibition
Yuanzheng Wang , Chen Zhang , Shuyan Han , Xiaoli Kong , Changyun Quan , Jun Wu , Wei Zhang

Recently, breast carcinoma has become a malignant tumor with the highest prevalence in women, and its recurrence and metastasis are the main causes of death [1,2]. In the early stages of breast carcinoma, traditional surgical resection and radiotherapy play an important role; however, the high incidence of recurrence and metastasis leads to a negative situation [3,4]. Chemotherapy is still an effective method to treat tumor metastasis. Nevertheless, direct use of chemotherapeutic drugs usually results in a low utilization rate and serious side effects, limiting the application of chemotherapeutic drugs [5]. Therefore, it is essential to develop novel therapeutic platforms to overcome the shortcomings of chemotherapy.
Drug delivery systems (DDSs) provide a potential method to improve drug availability and chemotherapeutic efficiency [6,7]. Hydrophobic drugs can be encapsulated in nanocarriers, which will improve their solubility and stability during circulation. Furthermore, functional drug carriers can achieve controlled drug release. This also improves drug metabolism with less leakage in circulation and more accumulation in the tumor site [8,9]. Various types of nano-carriers have been researched in DDS, including nanoparticles [10], micelles [11], liposomes [12], and nanogels [13] with different characteristics. Among them, nanogels (NGs) are important vehicles for chemotherapeutic drugs. The maximum retention space and porous structure of NGs are beneficial for drug loading, and they can also protect the drug from degradation and promote drug release in a controlled manner. Additionally, NG has nano-size for easy endocytosis, along with outstanding structural permeability, strength, high water absorption capacity, good dispersion stability, and fast response to external stimuli, showing high potential for drug delivery [14,15]. In particular, NGs based on biomacromolecules exhibit better properties in terms of biodegradability, biocompatibility, and low immunotoxicity [16].
Because of the poor efficiency of enhanced permeability and retention (EPR) effects and high interstitial pressure of solid tumors, passive targeting strategies usually have limited applications [17,18]. Active targeting may enhance therapeutic outcomes; however, its efficiency might be restricted by insufficient receptor density on the tumor surface [19,20]. Synthetic molecules can enhance tumor targeting, but they can easily induce immune responses and clearance by the reticuloendothelial system (RES) [21,22]. Therefore, a novel biomimetic cell membrane camouflage strategy has been envisioned. Compared with traditional modifications such as polyethylene glycol (PEG) layers, cell membranes derived from organisms have good biocompatibility, and more importantly, retain the biological properties of the original cells [23]. Erythrocyte, macrophage, platelet, stem cell, and various cancer cell membrane camouflaged nano-DDSs have been developed; these novel top-down coating strategies could prolong blood circulation time, reduce RES clearance, and accumulate DDSs in the lesion by retaining the surface antigenic protein from source cells [24]. Unlike erythrocyte cells, surface antigens on cancer cells can not only avoid RES phagocytosis but also promote the accumulation of DDSs in homologous tissue actively due to their homologous targeting ability [25].
Gelatin, a natural biological macromolecule, is a hydrolytic product of collagen [26]. Due to the presence of numerous functional groups on the molecular chain, gelatin can be used for the production of nanoparticles. Gelatin NGs have good biocompatibility and biodegradability, and their network structure provides a huge space for drug loading. However, the nonspecific recognition and RES clearance of gelatin NG-based DDSs limit their application in cancer therapy. Therefore, cell membrane camouflage on the surface has been evaluated as a feasible method. Studies have shown that after coating with an erythrocyte membrane, the phagocytosis of cisplatin (Pt) and methylene blue (MB) co-loaded gelatin NGs by macrophages was significantly inhibited. The tumor growth inhibition efficiency increased to 63.54% ± 8.82%, and the pulmonary metastasis inhibition rate increased to 97.75% ± 1.57% [27]. The natural occurrence and high tumor affinity of stem cells make the bone marrow-derived mesenchymal stem cell membrane camouflaged doxorubicin (DOX)-encapsulated gelatin NG accumulate in tumor tissue, thus effectively inhibiting solid tumor in vivo [28].
In this study, a 4T1 cancer cell membrane camouflaged biological gelatin NG with DOX loaded (M-NG@DOX) was developed for breast tumor therapy (Scheme 1). The gelatin NG was prepared by chemical cross-linking. DOX was encapsulated in the NG core (NG@DOX), and then the 4T1 cell membrane was modified on the surface through extrusion (M-NG@DOX). The results indicated that cancer cell membrane camouflaged NGs underwent less endocytosis by macrophages (RAW264.7) but were more internalized by 4T1 cells owing to homologous targeting. In the 4T1 xenograft mouse models, a much greater accumulation in tumor tissue was observed after injecting with M-NG, and the M-NG@DOX showed more effective tumor suppression efficiency and higher biosafety than free DOX did. These results indicated that the biomimetic cancer cell membrane-coated NG could be considered as a promising platform for chemotherapeutic drug delivery.
The aim of this study was to enhance the accumulation of the drug delivery system at the tumor site and prevent the clearance of RES, thereby improving the efficacy of tumor therapy. We applied camouflage of the 4T1 cell membrane onto the surface of gelatin NG and loaded DOX for tumor treatment. When the DDS was enveloped with the cancer cell membrane, it effectively evaded macrophage phagocytosis and extended the circulation time. The specific targeting of the 4T1 cancer cell membrane increased the uptake of DDS by 4T1 cells and enhanced accumulation at the tumor site, ultimately augmenting the effectiveness of cancer treatment in vivo.
First, a gelatin NG was synthesized using a two-step desolvation method, following a procedure previously reported in the literature [29]. The morphology of NG and M-NG was subsequently examined by transmission electron microscope (TEM). As illustrated in Fig. 1A, the TEM images reveal that the NGs were spherical particles, approximately 127 ± 18 nm in size. Upon repeated extrusion, the M-NG displayed a distinct core–shell structure, measuring around 140 ± 15 nm. The coating thickness was approximately 12 nm. This demonstrates the successful coating of the cancer cell membrane without altering the inherent NG structure. Moreover, the diameter of the NGs expanded from 313.0 ± 25.0 nm (polydispersity index (PDI): 0.061) to 340.5 ± 20.0 nm (PDI: 0.019) post membrane coating. Simultaneously, the zeta potential transitioned from −26.27 ± 0.59 mV to −13.20 ± 1.35 mV (Figs. 1B and C). This adjusted potential closely mirrored the surface potential of 4T1 vesicles, which stands at −7.99 ± 1.04 mV. Collectively, these findings solidify the effective preparation of M-NG. To ascertain the robustness of M-NG, its stability was tested through a dynamic light scattering (DLS) analysis, as presented in Fig. S1 (Supporting information). Upon immersing the M-NG in both phosphate buffered saline (PBS) and DMEM medium containing 10% fetal bovine serum (FBS) for a span of 7 days, no significant variations in aggregation or size were observed. This attests to M-NG's stability, proving its capacity to remain consistent in a physiological setting for, at the very least, a week.
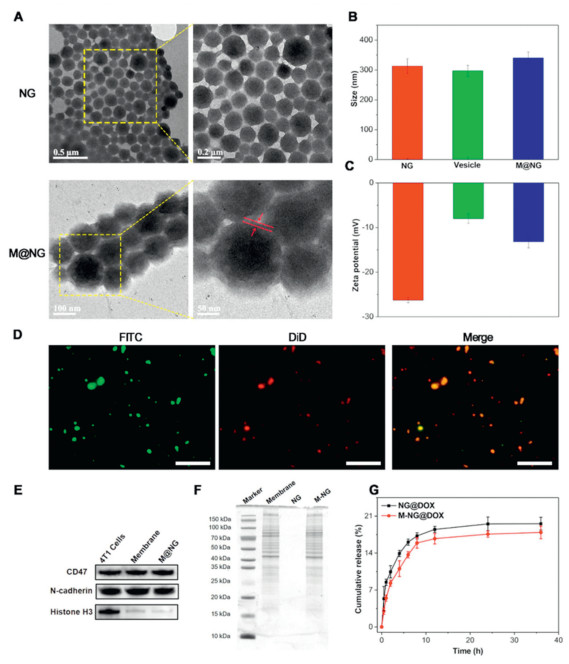
To validate the integrity of the core–shell structure of M-NG, the fluorescein isothiocyanate (FITC, green fluorescence) loaded NG core and DiD (red fluorescence) labeled 4T1 cell membrane were extruded following the standard protocol. The resultant co-labeled M-NG was then examined using an inverted fluorescence microscope. Fluorescence microscope images, as seen in Fig. 1D, revealed that a significant majority of M-NG simultaneously exhibited green fluorescent signals originating from the NG cores and red fluorescence signals from the 4T1 cell membrane shell, with a remarkable overlap. When considered alongside the TEM images and potential change, these observations underscore successful membrane fusion onto the NG surface.
In the next phase of our study, the protein content of M-NG was investigated via Western blot (WB) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analyses. Noteworthy among the membrane proteins are CD47 and N-cadherin. CD47 functions as a marker protein for 4T1 cells, pivotal in facilitating immune escape and anti-phagocytosis, while N-cadherin (pan-cadherin) plays a crucial role in promoting blood vessel proliferation, a factor indispensable for breast cancer invasion [30]. WB analysis findings illustrated that proteins CD47 and N-cadherin, present on the cancer cell surface, remained intact during M-NG preparation. Additionally, the nuclear marker protein, histone H3, was notably absent in both cell membrane vesicles and M-NG (Fig. 1E), further highlighting the effective capture of the cell membrane. An examination through SDS-PAGE (Fig. 1F) further corroborated these results, showcasing that, in comparison to the 4T1 cell membrane, most proteins persisted throughout the M-NG preparation process. In summation, the gathered evidence solidly affirms that the cancer cell membrane was adeptly coated onto the gelatin NG surface, all while preserving membrane protein integrity.
Owing to its excellent water absorption and intricate network structure, NGs emerge as an optimal choice for drug carriers. In our study, DOX was successfully encapsulated into the gelatin NG, leading to the formulations NG@DOX and M-NG@DOX. The ultraviolet–visible (UV–vis) spectra of both NG@DOX and M-NG@DOX exhibited a distinctive absorption peak around 480 nm (Fig. S2 in Supporting information), pointing to the efficient encapsulation of DOX. The encapsulation efficiency (EE) of DOX was recorded at 13.76% ± 1.78%, with its drug loading efficiency (DLE) hovering around 6.88% ± 0.36%.
When analyzing the in vitro drug release patterns of NG@DOX and M-NG@DOX in PBS (pH 7.4) at 37 ℃, it was observed that about 19.56% ± 1.21% of the drug was released from NG@DOX over 36 h (Fig. 1G). In contrast, roughly 17.90% ± 1.20% was released from M-NG@DOX. The slightly reduced drug release from M-NG@DOX can be associated with its diminished permeability, a consequence of the outer cell membrane coating on gelatin NG. Additionally, this membrane camouflage likely moderates the abrupt release tendencies of the drug-loaded platform.
A critical facet of biological defense, the clearance of foreign substances in organisms, primarily hinges on the potency of the immune system. Yet, the incessant proliferation of tumor cells induces antigen diversity, which in turn hinders their recognition and subsequent elimination by immune cells. CD47 stands as a prominent antigen on the 4T1 cell surface. Its capability to bind with signal regulatory protein α (SIRPα), a ligand found on macrophages, allows it to transmit a "don't eat me" directive, thereby inhibiting macrophage-mediated phagocytosis [31,32]. In our pursuit to gauge the immune evasion efficacy of M-NG, FITC-labeled M-NG was introduced to mouse monocyte macrophages (RAW264.7). Fig. 2A reveals a diminished green fluorescence in RAW264.7 cells exposed to M-NG when set against those treated with NG. This underscores a reduced uptake of M-NG by macrophages—a phenomenon likely stemming from the preservation of CD47 on the M-NG surface. Such findings accentuate the potential of the cancer cell membrane camouflage strategy in enhancing immune escape.
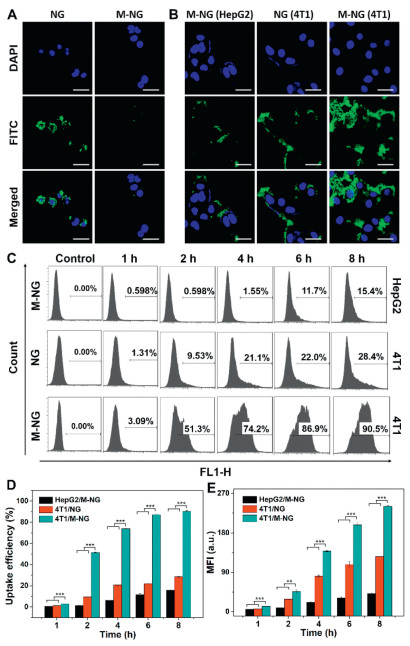
To delve deeper into the homologous targeting capability of M-NG, we evaluated the cellular uptake behaviors of FITC-labeled NG and M-NG against 4T1 and HepG2 cells, utilizing both CLSM and flow cytometry techniques. Fig. 2B illustrates that, post-treatment with M-NG, the green fluorescence intensity was markedly pronounced in the source 4T1 cells as opposed to the HepG2 cells. Concurrently, when exposed to M-NG, 4T1 cells exhibited heightened green fluorescence compared to their response to NG. This pattern signifies that modifications with the cancer cell membrane can amplify the targeting and internalization within source cells. It further elucidates that the unique homologous targeting characteristic is potent against heterotypic tumor cells. Flow cytometry further revealed a notable temporal dependency in the cellular uptake of either NG or M-NG. Evidently, 4T1 cells demonstrated the most efficient uptake of M-NG, as depicted in Fig. 2C. This uptake efficiency surged from 28.56% ± 0.38% to a striking 91.30% ± 0.63% post 4T1 cell membrane camouflage and after an 8-h incubation. This was also paired with the highest recorded average fluorescence intensity among the examined groups (Figs. 2D and E). Based on the observations above, it is apparent that the cancer cell membrane camouflage strategy bolsters the nanoplatform's homologous cell uptake efficiency, a feat achieved due to the presence of diverse antigens and inherent homotypic binding.
One of the foremost parameters when evaluating the performance of M-NG is its biocompatibility. The cytotoxic potential of M-NG was determined using the MTT assay. Both normal mouse mammary epithelial cells (HC11 cells) and mouse breast cancer cells (4T1 cells) were co-incubated with various concentrations of M-NG over a 24-h period. As evidenced in Fig. 3A, HC11 cells across all groups consistently exhibited high cell viability. Even at the peak concentration of 400 µg/mL, cell viability was sustained above 90%. In a similar vein, Fig. 3B reveals that 4T1 cell viability also exceeded 90%. Such observations validate the commendable biocompatibility of M-NG. Its minimal cytotoxic footprint suggests M-NG's potential as a safe avenue for drug delivery. Given that this delivery system is designed for intravenous administration, its blood compatibility is paramount. The hemolysis test reaffirmed this, demonstrating no significant hemolytic reactions with M-NG (Fig. S3 in Supporting information).
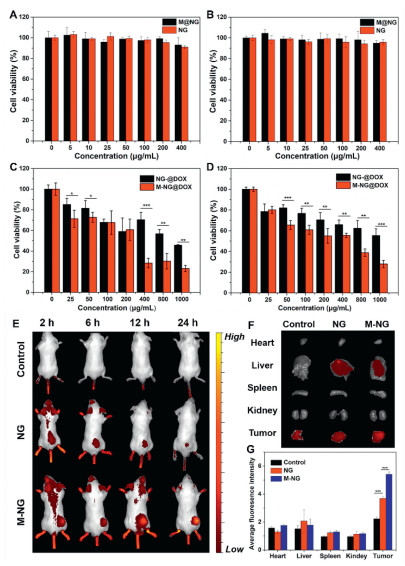
Leveraging its source cell-specific targeting prowess and elevated cell uptake efficiency, the in vitro therapeutic potency of M-NG@DOX against 4T1 cells was further scrutinized through the MTT assay. Fig. 3C depicts that M-NG@DOX outperformed NG@DOX in terms of cell-killing capability over a 24-h window. The cell viability plummeted from 55.39% ± 6.38% to a mere 27.94% ± 3.59% post cancer cell membrane camouflage, pointing towards the proficient uptake and internalization of M-NG@DOX by 4T1 cells. This variance in cytotoxicity became even more pronounced after 48 h, as illustrated in Fig. 3D. Here, the cell viability further dwindled to 23.09% ± 3.04% post M-NG@DOX treatment. To encapsulate, the precision targeting towards surface antigens amplified M-NG@DOX's cell uptake, culminating in enhanced antitumor effectiveness in vitro.
Having established the homologous targeting capability of M-NG in vitro, its in vivo specific binding attributes were next evaluated using the 4T1 cell xenograft mouse model. All animal experiments have been approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University (the approval number is SYSU-IACUC-2022-000281). Both Cy-7-labeled NG and M-NG were intravenously administered to 4T1 tumor-bearing mice and subsequently monitored with a Perkin Elmer in vivo imaging system. As showcased in the fluorescence snapshots of Fig. 3E, in comparison to free Cy-7 and NG, the Cy-7-labeled M-NG-treated groups exhibited heightened fluorescence intensity at the tumor locale post a 12-h incubation. This observation underscores its superior accumulation within the tumor region. This targeted accumulation was even more pronounced for the M-NG group after a 24-h span. Following this, major organs and tumors were extracted for ex vivo fluorescence imaging, as displayed in Fig. 3F. The fluorescence intensity within tumors treated with M-NG far surpassed other groups and organs. The average signal emanating from the fluorescence imaging was then quantified, as detailed in Fig. 3G. Notably, the tumor fluorescence intensity of M-NG was approximately 1.47 times that of pure NG. The marked distinction in aggregation degrees between NG and M-NG can be traced back to the biomimetic cancer cell membrane camouflage approach. The presence of various antigens on the membrane shell's surface, such as N-cadherin and CD47 (which was authenticated via WB analysis), facilitated specific adherence to the originating tumor tissue. Moreover, certain marker proteins like CD47 curbed RES clearance and macrophage-mediated phagocytosis, thereby extending the body's circulation duration. This, in turn, enhanced the tumor site accumulation of M-NG.
Building upon the promising homologous tumor accumulation observed in the 4T1 cell xenograft mouse model, we subsequently delved into assessing the antitumor efficacy of M-NG@DOX in solid tumors. The treatment schedule is elaborated in Fig. 4A. Mice underwent intravenous injections of PBS, free DOX, NG@DOX, or M-NG@DOX, each having a DOX dosage of 0.5 mg/kg. Over a 14-day treatment duration, therapeutic outcomes were closely monitored. Fig. 4B vividly highlights the stark differences: while tumors in the PBS-treated group proliferated swiftly, the counterparts treated with free DOX, NG@DOX, or M-NG@DOX manifested varying degrees of decelerated tumor growth. Post a 14-day regimen, the tumor growth inhibition (TGI) value for the free DOX, NG@DOX, and M-NG@DOX groups were charted at 19.14%, 52.79%, and 70.36% respectively, with M-NG@DOX championing the most potent tumor-inhibiting action. The 14th day post-treatment also saw tumors from each group being dissected, and as showcased in Fig. 4C, M-NG@DOX's exceptional in vivo antitumor performance was evident, boasting the smallest tumor volume in its group. Tumors from the NG@DOX cluster grew at a slower pace than those from the free DOX contingent. This can be attributed to the passive in vivo targeting capabilities and the modulated drug release properties inherent to nanostructures. The added advantage of a cancer membrane coating on M-NG@DOX ensures even better tumor inhibitory rates, likely due to the antigen's ability to specifically bind with 4T1 tumor tissue and dodge macrophage-mediated clearance.
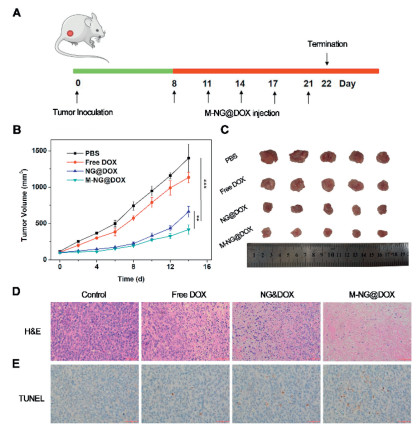
Diving deeper into the antitumor efficacy of M-NG@DOX, tumor slices, post a 14-day treatment, underwent hematoxylin and eosin (H&E) staining. Fig. 4D reveals that, in comparison to the control batch, a plethora of cytoplasm (colored red) dominated the experimental groups, a strong indication of nuclear degradation and cell apoptosis. Especially within the M-NG@DOX cohort, a pronounced presence of nuclear pyknosis and obliteration was evident, devoid of any dividing tumor cells in sight. Complementing this, the TUNEL staining of tumor sections, as captured in Fig. 4E, spotlighted the M-NG@DOX group with the highest brown nucleus percentage—an unequivocal testament to apoptosis. Such histopathological findings underscore the potential of M-NG@DOX to instigate peak necrosis within tumor tissues, achieving unparalleled in vivo antitumor efficacy largely by promoting tumor cell apoptosis.
Assessing the potential in vivo toxicity of M-NG@DOX is of paramount importance, particularly for therapies administered intravenously. Throughout the treatment duration, body weights were diligently recorded every alternate day, as visualized in Fig. S4 (Supporting information). Neither fatalities nor significant fluctuations in body weight were discerned in both the control and experimental sets, signifying the lack of adverse effects of NG@DOX and M-NG@DOX on the mice. Concurrently, the systemic toxicity of both NG@DOX and M-NG@DOX was further appraised using H&E staining of pivotal organs. As shown in Fig. S5 (Supporting information), no manifest tissue necrosis was detected across all organs, mirroring observations from the PBS cohort. Given the modest DOX dosage of 0.5 mg/kg, the free DOX group neither presented noteworthy weight deviations nor structural impairments within vital organs. Such compelling evidence of robust in vivo biocompatibility fortifies the foundation for biomimetic gelatin substances and the cell membrane coating approach, paving the way for its extended application in diverse biomedical arenas.
In summary, utilizing innovative techniques, we engineered a biomimetic gelatin NG camouflaged by a natural cancer cell membrane. This cutting-edge platform, created through a top-down assembly method, efficiently delivers DOX, offering an enhanced therapeutic approach against breast carcinoma. Through the successful fusion of cell membranes and the preservation of their native properties, M-NG not only zeroes in on source cancer cells but also augments cell uptake and internalization, culminating in improved therapeutic outcomes in vitro. Moreover, in vivo evaluations revealed M-NG's prowess in targeting tumor sites and its exceptional antitumor effects against mouse model xenografts. These benefits, coupled with minimal systemic side effects, can be credited to the homologous targeting prowess and immune evasion capabilities of the biomimetic cancer cell membrane "shell". Given these attributes, M-NG@DOX stands out as a potential drug delivery nanoplatform, primed to enhance targeting specificity and mitigate RES clearance, setting the stage for optimized carcinoma treatments.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by grants from Science and Technology Program of Guangzhou, China (No. 201804010146) and Guangzhou Science and Technology Program City-University Joint Funding Project (No. 2023A03J0001).
Supplementary material associated with this article can be found, in the online version, at doi:
M. Rozenblit, S. Mun, P. Soulos, et al., Breast Cancer Res. 23 (2021) 14. doi: 10.1186/s13058-021-01394-y
Y.Q. Xu, W.C. Chen, J. Liang, et al., J. Exp. Clin. Cancer Res. 40 (2021) 47. doi: 10.1186/s13046-020-01767-9
N.P. McAndrew, L. Bottalico, C. Mesaros, et al., NPJ Breast Cancer 7 (2021) 7. doi: 10.1038/s41523-020-00212-6
J.J. Cen, Y.P. Liang, Y. Huang, et al., Mol. Cancer 20 (2021) 19. doi: 10.1186/s12943-021-01314-w
C.X. Feng, Z.Y. Xiong, C. Wang, et al., Bioact. Mater. 6 (2021) 963–974.
D. Li, S. Liu, Y. Ma, et al., Small Methods 7 (2023) e2300204. doi: 10.1002/smtd.202300204
J. Chen, J. Yang, J. Ding, J. Mater. Chem. B 10 (2022) 7173–7182. doi: 10.1039/d2tb00581f
W.X. Zhou, Y.L. Zhang, S. Meng, et al., Small Methods 5 (2021) 2000920. doi: 10.1002/smtd.202000920
Y.L. Lo, C.H. Chang, C.S. Wang, et al., Theranostics 10 (2020) 6695–6714. doi: 10.7150/thno.45164
A.N. DuRoss, M.R. Landry, C.R. Thomas, et al., Cancer Lett. 500 (2021) 208–219. doi: 10.1016/j.canlet.2020.11.021
T. Zhang, B.H. Wu, O.U. Akakuru, et al., Cancer Lett. 500 (2021) 41–50. doi: 10.1016/j.canlet.2020.12.028
M. Hu, J. Zhang, L. Kong, et al., ACS Nano 15 (2021) 3123–3138. doi: 10.1021/acsnano.0c09681
M. Sun, L. He, Z. Fan, et al., Biomaterials 257 (2020) 120252. doi: 10.1016/j.biomaterials.2020.120252
X.R. You, L.Y. Wang, J.F. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107720. doi: 10.1016/j.cclet.2022.07.063
Y.Z. Ren, H.Q. Jing, Y. Zhou, et al., Chin. Chem. Lett. 34 (2023) 108161. doi: 10.1016/j.cclet.2023.108161
D.T. Nguyen, V.T. Dinh, L.H. Dang, et al., Polymers 11 (2019) 814. doi: 10.3390/polym11050814
J. Wang, H. Pan, J.Y. Li, et al., Chin. Chem. Lett. 34 (2023) 107828. doi: 10.1016/j.cclet.2022.107828
H. Li, J.B. Lu, C.Q. Yan, L.W. Xu, Int. J. Polym. Mater. Polym. Biomater. 69 (2020) 1157–1166. doi: 10.1080/00914037.2019.1667804
H.L. Zhao, N. Li, C.X. Ma, et al., Chin. Chem. Lett. 34 (2023) 107699. doi: 10.1016/j.cclet.2022.07.042
H. Zhao, P. Zhou, K. Huang, et al., Adv. Healthc. Mater. 7 (2018) 1800296. doi: 10.1002/adhm.201800296
X.R. Chen, B.B. Liu, R.L. Tong, et al., Biomater. Sci. 9 (2021) 590–625. doi: 10.1039/d0bm01617a
Y.L. Chen, K. Cheng, Nano Res. 13 (2020) 2617–2624. doi: 10.1007/s12274-020-2931-5
J.B. Gao, F. Wang, S.H. Wang, et al., Adv. Sci. 7 (2020) 1903642. doi: 10.1002/advs.201903642
Z.F. Ma, S. Liu, Y. Ke, et al., Biomaterials 255 (2020) 120141. doi: 10.1016/j.biomaterials.2020.120141
R.L. Wang, H. Yang, R.X. Fu, et al., Cancers 12 (2020) 3136. doi: 10.3390/cancers12113136
C.Q. Fan, K.G. Xu, Y. Huang, et al., Bioact. Mater. 6 (2021) 4161–4162.
Y.H. Zhai, W. Ran, J.H. Su, et al., Adv. Mater. 30 (2018) 1802378. doi: 10.1002/adma.201802378
C.Y. Gao, Z.H. Lin, B. Jurado-Sánchez, et al., Small 12 (2016) 4056–4062. doi: 10.1002/smll.201600624
L. Wang, L. Chen, J. Wang, et al., Chin. Chem. Lett. 33 (2022) 1956–1962. doi: 10.1016/j.cclet.2021.10.070
S.Y. Li, H. Cheng, W.X. Qiu, et al., Biomaterials 142 (2017) 149–161. doi: 10.1016/j.biomaterials.2017.07.026
H.P. Sun, J.H. Su, Q.S. Meng, et al., Adv. Mater. 28 (2016) 9581. doi: 10.1002/adma.201602173
S. Liu, Z. Wang, Q. Wei, et al., J. Control. Release 363 (2023) 721–732. doi: 10.1016/j.jconrel.2023.09.039

Scheme 1 Preparation of 4T1 cell membrane camouflaged biomimetic nanoplatform M-NG@DOX with homologous targeting ability for cancer therapy.

Figure 1 Characterizations of M-NG. (A) TEM images of NG and M-NG morphologies. (B) DLS data for NG and M-NG (n = 3). (C) Zeta potential values associated with NG and M-NG (n = 3). (D) Inverted fluorescence microscope images of M-NG, highlighting the co-localization between gelatin NG (FITC, green) and 4T1 cell membranes (DiD, red). Scale bar: 10 µm. (E) Assessment of CD47, N-cadherin, and histone H3 proteins in 4T1 cells, 4T1 cell membrane vesicles, and M-NG using WB. (F) SDS-PAGE protein profiles for 4T1 cell membrane, NG, and M-NG. (G) Observation of cumulative DOX release patterns from NG@DOX and M-NG@DOX at pH 7.4 and 37 ℃ in vitro (n = 3). Values were noted as the mean ± standard deviation (SD).

Figure 2 Assessment of phagocytosis of M-NG by macrophages and the evaluation of homologous targeted uptake of M-NG in vitro. (A) Uptake by RAW264.7 of FITC-labeled NG and M-NG following a 4-hour incubation period. Scale bar: 30 µm. (B) Comparative cellular uptake: M-NG in HepG2 cells, NG in 4T1 cells, and M-NG in 4T1 cells, visualized using CLSM. Scale bar: 30 µm. (C) Flow cytometry analysis of the cellular uptake against various cells lines with corresponding cellular uptake efficiency (D) and mean fluorescence intensity (MFI) values (E) qualified (n = 3). **P < 0.01, *** P < 0.001. Values were noted as the mean ± SD.

Figure 3 Evaluation of the in vitro antitumor efficacy and in vivo biodistribution of M-NG. (A) Viability of HC11 cells post a 24-h incubation with NG and M-NG (n = 3). (B) Viability of 4T1 cells following a 24-h incubation with NG and M-NG (n = 3). Viability of 4T1 cells post treatment with NG or M-NG for durations of 24 h (C) and 48 h (D) (n = 3). (E) Visual representation of the biodistribution of free Cy-7, Cy-7-NG or Cy-7-M-NG in the whole body. (F) Ex vivo imaging of major organs and tumors at 24 h post-injection with comparative fluorescence intensity of Cy-7 across major organs and tumors. (G) Quantitative analysis of average fluorescence imaging signals in vivo (n = 3). * P < 0.5, ** P < 0.01, *** P < 0.001. Values were noted as the mean ± SD.

Figure 4 Illustration of the antitumor efficacy of M-NG@DOX in vivo. (A) Schematic representation of the study's timeline. (B) Growth curves of xenograft tumors in mice (n = 5). Values were noted as the mean ± SD. ** P < 0.01, *** P < 0.001. (C) Representative images of xenograft tumors across different experimental groups. (D) H&E and (E) TUNEL stained sections of tumor tissues post a 14-day treatment duration. Scale bar: 100 µm.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: