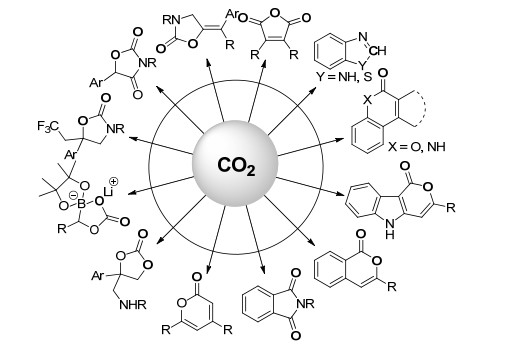
 图 1
二氧化碳参与的环化新反应合成的杂环化合物
Figure Figure1.
Heterocyclic compounds from new cyclization reactions using carbon dioxide
图 1
二氧化碳参与的环化新反应合成的杂环化合物
Figure Figure1.
Heterocyclic compounds from new cyclization reactions using carbon dioxide
引用本文:
张文珍, 张宁, 郭春晓, 吕小兵. 二氧化碳参与的环化反应最新研究进展[J]. 有机化学,
2017, 37(6): 1309-1321.
doi:
10.6023/cjoc201701031

Citation: Zhang Wenzhen, Zhang Ning, Guo Chunxiao, Lü Xiaobing. Recent Progress in the Cyclization Reactions Using Carbon Dioxide[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1309-1321. doi: 10.6023/cjoc201701031
Citation: Zhang Wenzhen, Zhang Ning, Guo Chunxiao, Lü Xiaobing. Recent Progress in the Cyclization Reactions Using Carbon Dioxide[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1309-1321. doi: 10.6023/cjoc201701031
二氧化碳参与的环化反应最新研究进展
English
Recent Progress in the Cyclization Reactions Using Carbon Dioxide
Abstract:
Carbon dioxide is a cheap, abundant and renewable C1 feedstock. Methodology study on the transformation of carbon dioxide into highly value-added chemicals has become one of the most active topics in organic chemistry. Owing to the diversity of cyclization reaction and vast occurrence of various heterocyclic motifs in biologically important molecules, the cyclization reactions using carbon dioxide have gained much attention. This review therefore aims to principally describe the recent progress in the new cyclization reactions using carbon dioxide as feedstock to synthesize lactams, lactones, cyclic anhydrides, benzothiazoles, benzimidazoles and other heterocyclic compounds.
-
Key words:
- carbon dioxide
- / cyclization reaction
- / green chemistry
- / organic synthesis
- / heterocycles
-
二氧化碳作为储量丰富、廉价易得的可再生碳一资源其催化转化得到了化学工作者的广泛关注[1~5].将二氧化碳高效转化成高附加值化学品的有机合成新方法学研究已经成为目前最为活跃的研究方向之一.虽然二氧化碳固有的热力学及动力学稳定性限制了它作为碳一合成子的广泛应用, 但已初步建立起来的多种二氧化碳新有机反应体系可将二氧化碳成功转化成在制药等精细化工中有着重要用途的合成中间体[6~11].由于环化反应种类的多样性以及环化产物在众多生物活性分子结构中的广谱性, 二氧化碳参与的环化反应也深受广大研究者重视[12, 13].目前二氧化碳与环氧烷烃或炔丙醇反应合成环状碳酸酯、与氮杂环丙烷或炔丙胺反应合成噁唑啉酮、与邻氨基苯腈反应合成喹唑啉二酮这三大类反应的新催化体系研究较多, 已有很多综述从不同角度做了概括总结[12~18], 本文将不再主要介绍.最近发展起来的二氧化碳参与的其他多种环化新反应可高效合成多种杂环化合物.如在非还原性条件下, 二氧化碳可通过内酰胺化、内酯化、环加成等反应合成内酰胺、内酯、邻苯二甲酰亚胺、环状酸酐等(图 1); 在硅烷等还原剂的存在下, 邻苯二胺或邻氨基苯硫醇可与二氧化碳进行还原性羧化环化反应得到苯并咪唑或苯并噻唑类化合物.以上环化反应在建立起新的二氧化碳活化转化模式的同时, 也为制备杂环化合物提供了更为绿色的合成路线.本文按照产物的种类、反应底物类型分层次对最近二氧化碳参与的环化新反应及其机理进行综述, 同时也介绍了一些二氧化碳作为反应底物但不参与成环过程合成环状羧酸的新反应.
图 1
二氧化碳参与的环化新反应合成的杂环化合物
Figure Figure1.
Heterocyclic compounds from new cyclization reactions using carbon dioxide
1 合成内酰胺类化合物
通过sp2碳氢键的二氧化碳羧化反应合成内酰胺最近取得了重要的研究进展.相比于端炔烃中sp碳氢键的二氧化碳羧化反应, 芳烃或烯烃中sp2碳氢键和烷烃中sp3碳氢键的二氧化碳羧化反应条件较为苛刻[19, 20].在芳烃或烯烃底物中引入氨基等官能团, 通过形成内酰胺等环状化合物的sp2碳氢键二氧化碳羧化反应在热力学上更有利.
2016年初, 余达刚课题组[21]报道了二氧化碳参与的邻烯基/杂环芳基苯胺中sp2碳氢键的内酰胺化反应(Scheme 1).仅在叔丁醇钠作为碱, 二乙二醇二甲醚为溶剂, 140 ℃的反应条件下, 一系列邻烯基/杂环芳基苯胺化合物1与101 kPa的二氧化碳进行sp2碳氢键的内酰胺化反应, 以较高收率得到2-喹啉酮类化合物2.此反应底物适用性广, 产物易于衍生化, 操作简便, 易于放大, 可用于构建多种重要药物分子(如2c)及重要前体(如2a和2d) (Scheme 1).作者首次明确提出了“CO2=CO+O (氧化剂)”的概念[22].相比于传统的一氧化碳作为羰基源的钯催化邻烯基/杂环芳基苯胺sp2碳氢键的氧化羰基化反应, 二氧化碳为羰基源的内酰胺化反应在无过渡金属参与且氧化还原中性的条件下进行, 对环境友好, 也更容易在精细化学品工业中应用.机理研究表明在此反应体系中可能生成高活性的邻烯基/杂环芳基苯基异腈酸酯中间体3, 然后异构化为内酰胺产物(Scheme 1).
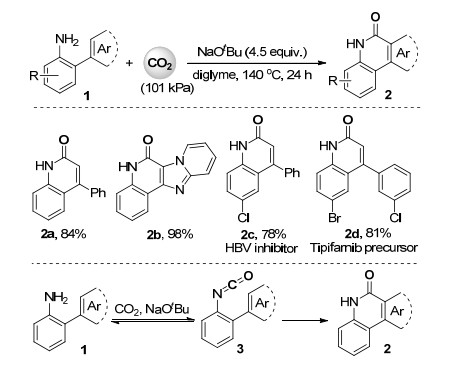 图式1
二氧化碳参与的邻烯基/芳基苯胺中sp2碳氢键的内酰胺化反应
Scheme1.
Lactamization of sp2-C—H bonds in 2-alkenyl and 2-aryl anilines with carbon dioxide
图式1
二氧化碳参与的邻烯基/芳基苯胺中sp2碳氢键的内酰胺化反应
Scheme1.
Lactamization of sp2-C—H bonds in 2-alkenyl and 2-aryl anilines with carbon dioxide
随后, 席婵娟课题组[23]报道了无金属参与的邻芳基苯胺中sp2碳氢键与二氧化碳的内酰胺化反应(Scheme 2).在1, 5, 7-三氮杂二环[4.4.0]癸-5-烯(TBD)和三氟甲磺酸甲酯(MeOTf)的共同作用下, 一系列邻苯基/杂环芳基苯胺化合物4与101 kPa的二氧化碳在邻二氯苯中于140 ℃反应12 h, 以较高收率得到相应的内酰胺产品5.上述叔丁醇钠反应系统中不适用的邻苯基苯胺底物在此TBD/MeOTf体系中能顺利进行sp2碳氢键的内酰胺化反应.反应收率主要受底物电子效应的影响, 富电子的邻芳基苯胺化合物反应活性更高.经过系统的对照实验, 作者给出了可能的反应机理(Scheme 2), 邻芳基苯胺首先与TBD二氧化碳加合物[24]及1 equiv.的MeOTf反应生成氨基甲酸酯6, 随后6在高温下脱去甲醇生成的邻芳基苯基异氰酸酯(7)未直接进行环化反应, 而是先和过量的MeOTf反应得到高活性的反应中间体8后再发生环化反应得到6-甲氧基菲啶中间体9, 随后9与上一步产生的三氟甲磺酸反应得到2-喹啉酮产物.
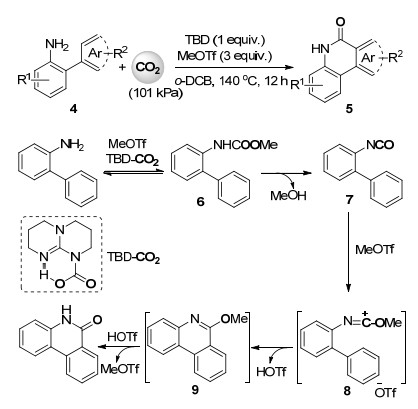 图式2
无金属参与的邻芳基苯胺中sp2-碳氢键与二氧化碳的内酰胺化反应
Scheme2.
Metal-free lactamization of sp2-C—H bonds in 2-arylanilines with carbon dioxide
图式2
无金属参与的邻芳基苯胺中sp2-碳氢键与二氧化碳的内酰胺化反应
Scheme2.
Metal-free lactamization of sp2-C—H bonds in 2-arylanilines with carbon dioxide
最近, 成江课题组[25]报道了钯催化邻碘苯胺(10)、N-对甲基苯磺酰腙11和101 kPa的二氧化碳的多组分反应(Scheme 3), 高效合成了一系列4-芳基-2-喹啉酮化合物12.此多组分反应形成了包含两组碳碳单键, 一组碳碳双键和一组碳氮单键共四组新的化学键.作者推测的反应机理主要为邻碘苯胺与N-对甲基苯磺酰腙在叔丁醇钠作为碱及钯催化下通过钯卡宾的迁移插入形成邻烯基苯胺化合物13 (Scheme 3), 然后再与二氧化碳反应经异氰酸酯中间体得到内酰胺产物.由于在最优反应条件下邻碘苯基异氰酸酯可与N-对甲基苯磺酰腙反应得到4-芳基-2-喹啉酮产物, 作者也未排除邻碘苯胺先于二氧化碳反应生成邻碘苯基异氰酸酯, 然后再与N-对甲基苯磺酰腙进行迁移插入/内酰胺化这一反应途径(Scheme 3).
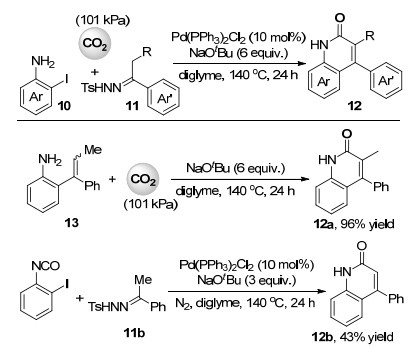 图式3
钯催化邻碘苯胺、N-对甲基苯磺酰腙和二氧化碳的多组分反应
Scheme3.
Palladium-catalyzed multi-component reactions of 2-iodoanilines, N-tosylhydrazones and carbon dioxide
图式3
钯催化邻碘苯胺、N-对甲基苯磺酰腙和二氧化碳的多组分反应
Scheme3.
Palladium-catalyzed multi-component reactions of 2-iodoanilines, N-tosylhydrazones and carbon dioxide
2 合成内酯类化合物
2.1 sp2碳氢键内酯化反应
2013年, Iwasawa课题组[26]报道了二价钯催化2-羟基苯乙烯类化合物中烯基sp2碳氢键与二氧化碳的直接羧化反应(Scheme 4).在5 mol%醋酸钯为催化剂, 3 equiv.的碳酸铯作为碱, 二乙二醇二甲醚为溶剂, 100 ℃的最优反应条件下, 包含给电子、吸电子或杂环取代基的一系列2-羟基苯乙烯化合物14与101 kPa的二氧化碳进行sp2碳氢键内酯化反应, 以中等到较高收率得到相应的香豆素类化合物15.反应机理研究表明底物14a首先与醋酸钯和碳酸铯反应生成烯基环钯配合物16, 当量反应分离得到了此配合物, X射线单晶衍射也确认了其分子结构.随后进行的二氧化碳羧化配合物16中碳钯键形成羧基钯中间体17的过程虽然是可逆的, 对照实验也证明17很容易脱羧直接回到热力学上更稳定的配合物16, 但反应过程中17可与另外一分子的反应底物和碳酸铯作用, 释放出产物内酯的同时再生反应活性中间体16, 使反应朝着羧化方向进行, 完成催化循环(Scheme 4).
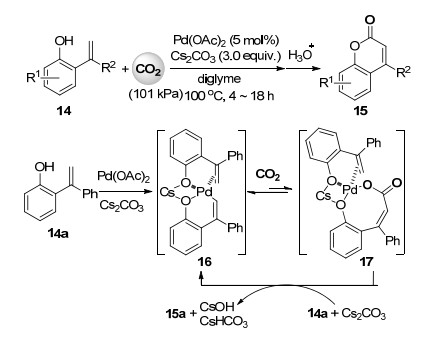 图式4
钯催化邻羟基苯乙烯sp2-碳氢键与二氧化碳的内酯化反应
Scheme4.
Palladium-catalyzed lactonization of sp2-C—H bonds in 2-hydroxystyrenes with carbon dioxide
图式4
钯催化邻羟基苯乙烯sp2-碳氢键与二氧化碳的内酯化反应
Scheme4.
Palladium-catalyzed lactonization of sp2-C—H bonds in 2-hydroxystyrenes with carbon dioxide
最近, 余达刚课题组[27]在之前sp2碳氢键的内酰胺化反应的基础上, 发展了无过渡金属参与的邻杂环芳基苯酚及邻羟基苯乙烯中sp2碳氢键与二氧化碳的内酯化反应(Scheme 5).一系列邻咪唑并[1, 2-a]吡啶基苯酚18在叔丁醇钾作为碱时与101 kPa的二氧化碳进行sp2碳氢键内酯化反应, 高收率地得到并四环结构的内酯化合物19.作者推测的反应机理为底物18首先与叔丁醇钾反应得到酚钾20后, 连续与两分子二氧化碳反应得到中间体22, 随后分子内亲核进攻得到内酯产物19 (Scheme 5).此无过渡金属参与的反应体系在N, N-二甲基甲酰胺(DMF)作为溶剂时也适用于富电子的邻羟基苯乙烯底物中sp2碳氢键与二氧化碳的内酯化反应合成香豆素类化合物15 (Scheme 5).
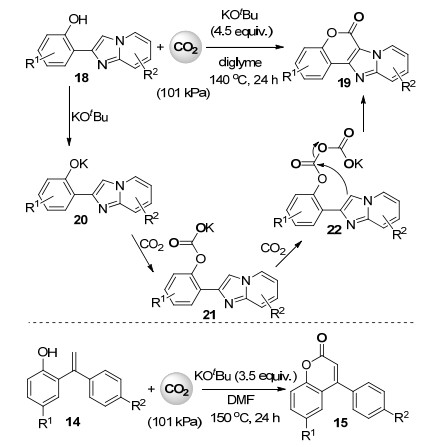 图式5
无过渡金属参与邻杂环芳基苯酚及邻羟基苯乙烯中sp2-碳氢键与二氧化碳的内酯化反应
Scheme5.
Transition-metal-free lactonization of sp2-C—H bonds in 2-arylphenols/ 2-hydroxystyrenes with carbon dioxide
图式5
无过渡金属参与邻杂环芳基苯酚及邻羟基苯乙烯中sp2-碳氢键与二氧化碳的内酯化反应
Scheme5.
Transition-metal-free lactonization of sp2-C—H bonds in 2-arylphenols/ 2-hydroxystyrenes with carbon dioxide
2015年, Skrydstrup课题组[28]报道了无金属参与的, TBD催化2-炔基吲哚类化合物23与二氧化碳的羧化环化反应高收率合成并三环的内酯化合物24 (Scheme 6).当R2为芳基时, 反应只需使用0.3 equiv.的TBD就可高效进行.当R1为烷基取代时底物的反应活性较差, 需要将TBD的用量提高到1 equiv.通过一系列对照实验作者提出了可能的反应机理(Scheme 6).首先底物23中吲哚环上sp2碳氢键被TBD二氧化碳加合物羧化形成中间体25, 然后25重新芳构化后得到的2-炔基-3-羧基吲哚中间体26进行6-endo-dig型分子内环化反应得到内酯产物24.
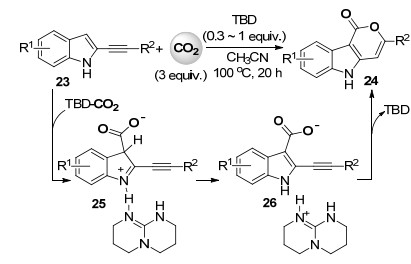 图式6
TBD催化2-炔基吲哚与二氧化碳的羧化环化反应
Scheme6.
TBD-catalyzed carboxylative cyclization reactions of 2-alkynyl indoles with carbon dioxide
图式6
TBD催化2-炔基吲哚与二氧化碳的羧化环化反应
Scheme6.
TBD-catalyzed carboxylative cyclization reactions of 2-alkynyl indoles with carbon dioxide
2.2 芳炔参与的多组分环化反应
芳炔作为高活性的反应中间体已广泛应用于多组分有机合成反应中[29].芳炔通常表现出很高的亲电性, 容易与亲核试剂先反应得到芳基负离子中间体, 然后再与各种亲电试剂反应得到1, 2-二取代的芳烃.早在2006年已有芳炔与亚胺及二氧化碳的三组分环化反应报道[30]. 2014年, Kobayashi课题组[31]报道了氮杂环卡宾铜配合物催化的芳炔、端炔和二氧化碳的多组分环化反应以中等到较高收率合成异香豆素类化合物28(Scheme 7).此反应体系采用碘化亚铜与膦配体作为催化剂时, 不但羧化产物收率低, 5-exo-dig和6-endo-dig型环化产物的选择性也很差.当使用氮杂环卡宾铜配合物作为催化剂, 在乙腈和四氢呋喃的混合溶剂中反应可高收率高选择性地得到6-endo-dig型环化产物.结合已有的报道及对照实验, 作者提出的催化反应机理见Scheme 7.端炔与铜配合物首先在碳酸铯作用下形成炔铜配合物.虽然我们课题组报道了二氧化碳可以插入此炔铜配合物的Csp—Cu键形成炔酸铜化合物[32], 但在此反应中作者未观察到相关产物.芳炔前体27在CsF作用下形成芳炔后插入炔铜配合物形成邻炔基芳基铜中间体29, 二氧化碳插入29中的碳铜键得到邻炔基芳甲酸铜中间体30后进行6-endo-dig型分子内环化反应形成内酯配合物31.由于使用氘代的端炔反应时异香豆素产物中没有检测到氘原子的引入, 所以作者认为铜配合物催化剂的再生和释放出产品的过程不是通过31的质子化, 而是通过31与铯盐的转金属化反应实现的(Scheme 7).
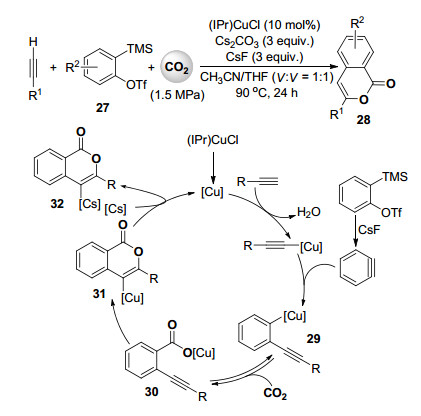 图式7
铜催化芳炔、端炔和二氧化碳的多组分羧化环化反应
Scheme7.
Copper-catalyzed multi-component carboxylative cyclization reactions of arynes, terminal alkynes and carbon dioxide
图式7
铜催化芳炔、端炔和二氧化碳的多组分羧化环化反应
Scheme7.
Copper-catalyzed multi-component carboxylative cyclization reactions of arynes, terminal alkynes and carbon dioxide
2.3 芳香酮参与的二氧化碳羧化/环化反应
众所周知, 在适当碱存在下, 酮羰基α位碳氢键可经烯醇式中间体与二氧化碳进行羧化反应得到β酮酸[33].基于此反应, 许多炔基酮与二氧化碳的羧化环化反应已被报道[34~36]. 2015年, 我们课题组[37]发展了1, 8-二氮杂二环十一碳-7-烯(DBU)促进的邻羟基苯乙酮33及邻乙酰胺基苯乙酮34与3 MPa的二氧化碳的羧化环化反应(Scheme 8), 高选择性地合成了一系列含有不同取代基的4-羟基香豆素类化合物35和4-羟基-2-喹啉酮类化合物36.其中在邻乙酰胺基苯乙酮与二氧化碳反应中酰基发生了迁移.
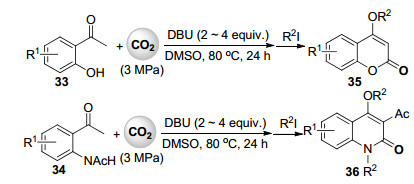 图式8
DBU促进的邻羟基/乙酰胺基苯乙酮与二氧化碳的羧化环化反应
Scheme8.
DBU-promoted carboxylative cyclization reactions of 2-hydroxy and 2-acetamidoacetophenone with carbon dioxide
图式8
DBU促进的邻羟基/乙酰胺基苯乙酮与二氧化碳的羧化环化反应
Scheme8.
DBU-promoted carboxylative cyclization reactions of 2-hydroxy and 2-acetamidoacetophenone with carbon dioxide
基于上述反应, 2016年我们课题组[38]发展了氟化铯促进的丙烯基酮37经γ位碳氢键与3 MPa的二氧化碳的羧化环化反应(Scheme 9), 以中等至较高收率合成了包含芳基、烷基及杂环等取代基的2-吡喃酮化合物38.通过18O标记的二氧化碳实验发现, 2-吡喃酮产物中只有一个氧原子来自于二氧化碳.依次推测可能的反应机理是丙烯基酮37在碱作用下首先形成共轭结构的烯醇式中间体39, 然后经γ位与二氧化碳羧化反应后形成羧基中间体40, 在过量碱的作用下, 40进一步脱质子后再通过烯醇式氧原子对羧基亲核进攻完成分子内酯化反应得到2-吡喃酮产物(Scheme 9).此反应也可能通过中间体39与两分子的二氧化碳反应形成类似于Scheme 5中中间体22的中间体41, 经分子内亲核进攻得到内酯产物.丙烯基酮37自身二聚的副反应通过提高二氧化碳压力和降低底物浓度得到了有效抑制.
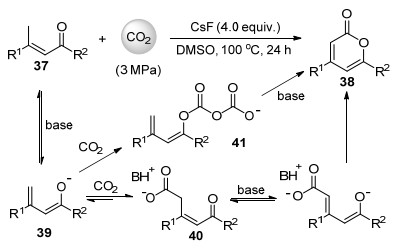 图式9
丙烯基酮与二氧化碳的羧化环化反应
Scheme9.
Carboxylative cyclization reactions of substituted propenyl ketones with carbon dioxide
图式9
丙烯基酮与二氧化碳的羧化环化反应
Scheme9.
Carboxylative cyclization reactions of substituted propenyl ketones with carbon dioxide
2015年, Murakami课题组[39]报道了在紫外甚至可见光驱动下, 邻烷基苯基酮化合物42在室温下与101 kPa的二氧化碳进行羧化反应高收率合成了邻酰基苯乙酸化合物43(Scheme 10).作者推测的反应机理中受光激发后的邻烷基苯基酮42中酮羰基攫取苯环邻位甲基上的氢后先形成1, 4-双自由基中间体44后, 立刻产生邻苯碳醌中间体45, 其中顺式构型的45通过1, 5-氢迁移回到原料43, 而反式构型的45与二氧化碳进行[4+2]环加成反应得到不稳定的羟基内酯产物46后, 经开环异构化得到邻酰基苯乙酸产物43 (Scheme 10).虽然此反应从表观上看不属于环化反应, 但作者认为反应过程中包含二氧化碳参与的[4+2]环加成形成内酯的过程.
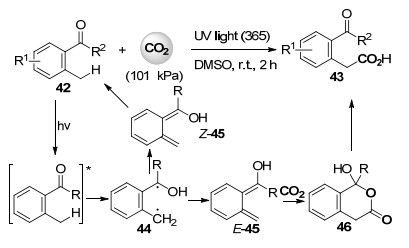 图式10
光驱动邻烷基苯基酮与二氧化碳的羧化反应
Scheme10.
Light-driven carboxylation of 2-alkylphenyl ketones with carbon dioxide
图式10
光驱动邻烷基苯基酮与二氧化碳的羧化反应
Scheme10.
Light-driven carboxylation of 2-alkylphenyl ketones with carbon dioxide
3 合成邻苯二甲酰亚胺
2014年, Biju课题组[40]报道了芳炔、异腈和101 kPa的二氧化碳的多组分环化反应合成邻苯二甲酰亚胺类化合物47 (Scheme 11).作者推测的反应机理见Scheme 11.由于异腈具有较高的亲核反应活性, 其亲核进攻原位形成的芳炔中间体形成1, 3-两性离子中间体后再亲核进攻二氧化碳得到中间体48, 羧基氧分子内亲核进攻闭环得到亚胺基异苯并呋喃酮49.随后在氟离子的作用下, 49开环后再次闭环形成热力学上更稳定的邻苯二甲酰亚胺产物(Scheme 11).
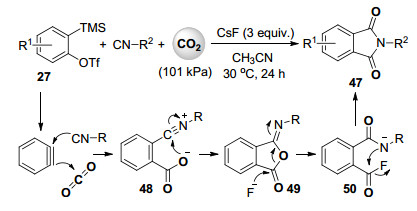 图式11
芳炔、异腈和二氧化碳的多组分羧化环化反应
Scheme11.
Multi-component carboxylative cyclization reactions of arynes, isocyanides and carbon dioxide
图式11
芳炔、异腈和二氧化碳的多组分羧化环化反应
Scheme11.
Multi-component carboxylative cyclization reactions of arynes, isocyanides and carbon dioxide
4 合成环状酸酐
2014年, Sakaki和Tsuji课题组[41]报道了镍催化内炔与101 kPa二氧化碳的双羧化反应, 高收率合成了取代的马来酸酐化合物51 (Scheme 12).反应体系中联吡啶作为配体, 锌粉作为还原剂, 溴化镁作为添加剂对酸酐产物的形成缺一不可.
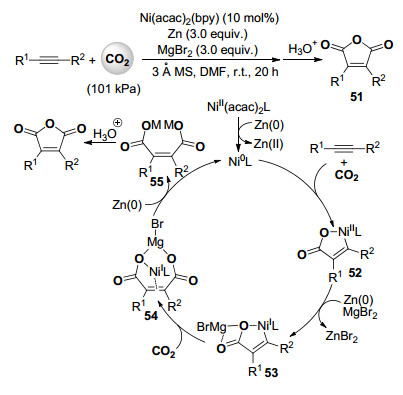 图式12
镍催化内炔与二氧化碳的双羧化反应合成马来酸酐
Scheme12.
Nickel-catalyzed double carboxylations of internal alkynes with carbon dioxide to synthesize maleic anhydrides
图式12
镍催化内炔与二氧化碳的双羧化反应合成马来酸酐
Scheme12.
Nickel-catalyzed double carboxylations of internal alkynes with carbon dioxide to synthesize maleic anhydrides
通过密度泛函理论计算及对照实验, 作者推测此反应的机理包括乙酰丙酮镍配合物首先被锌粉还原为高活性的零价镍配合物后, 与内炔底物和一分子的二氧化碳进行氧化加成得到经典的镍杂五元环内酯配合物52, 接着在锌粉和溴化镁的共同作用下, 二价镍配合物52被还原成一价镍配合物53, 其中与镁原子成离子键的羧基也配位在镍原子上.随后第二分子的二氧化碳插入碳镍键后形成中间体54, 在锌粉的作用下, 54中的一价镍被还原成零价镍后继续进入下一轮催化循环, 释放出的马来酸盐55经酸后处理形成马来酸酐产物(Scheme 12). 密度泛函理论计算表明, 溴化镁在将二价镍配合物52被还原成一价镍配合物53和第二分子二氧化碳插入过程中起关键的促进作用.
 图式13
钯催化炔丙胺, 芳基碘和二氧化碳的多组分羧化环化反应
Scheme13.
Palladium-catalyzed multi-component carboxylative cyclization reactions of propargylamines, aryl iodides and carbon dioxide
图式13
钯催化炔丙胺, 芳基碘和二氧化碳的多组分羧化环化反应
Scheme13.
Palladium-catalyzed multi-component carboxylative cyclization reactions of propargylamines, aryl iodides and carbon dioxide
5 合成噁唑啉酮
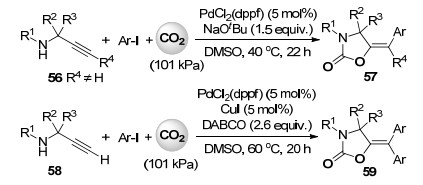
二氧化碳与与氮杂环丙烷或炔丙胺反应合成噁唑啉酮是目前二氧化碳转化研究中最常见的一类重要反应[14~18].最近研究者也发展了一些新的二氧化碳参与的环化反应合成噁唑啉酮.
2016年, Nevado课题组[42]报道了钯催化炔丙胺, 芳基碘和101 kPa二氧化碳的多组分羧化环化反应(Scheme 13), 以较高产率合成噁唑啉酮类化合物.与传统的路易斯酸催化的炔丙胺与二氧化碳羧化环化反应不同的是, 此反应中利用零价钯与芳基碘氧化加成后得到的二价钯配合物作为路易斯酸活化炔丙胺56中的碳碳叁键, 在促进羧化环化反应形成噁唑啉酮环后, 再经还原消除过程在双键上引入芳基, 得到最终的产物57.当使用端炔丙胺底物58时, 只需使用过量的芳基碘代物和添加5 mol%的碘化亚铜, 将炔丙胺56反应体系中的碱叔丁醇钠换成三乙烯二胺(DABCO), 便可在钯催化下先进行Sonogashira偶联, 然后再进行多组分羧化环化反应得到双键端位上引入两个相同芳基的噁唑啉酮产物59 (Scheme 13).
2016年, 余达刚课题组[43]首次实现了铜催化的二氧化碳参与的烯丙胺选择性氧三氟甲基化反应(Scheme 14).在10 mol% [Cu(MeCN)4]PF6为催化剂, 2 equiv.的DBU为碱, 一系列烯丙胺化合物60与101 kPa的二氧化碳和Togni试剂在室温下进行反应, 高收率, 高化学、区域及非对映异构体选择性地合成了一系列具有重要生理活性的含三氟甲基官能团的噁唑啉酮类化合物61.此反应条件温和、操作简便、可进行克级规模制备、产物分子重要且易于衍生化.通过一系列的对照实验及单一非对映异构体螺环产物61a的晶体结构分析, 作者推测的反应机理见Scheme 14.首先一价铜配合物与Togni试剂之间的单电子转移产生三氟甲基自由基的同时生成二价铜配合物, 同时60a和二氧化碳反应得到的氨基甲酸化合物62与此二价铜配合物反应形成63, 其中二氧化碳与氨基反应形成氨基甲酸酯的过程有效地抑制了氮三氟甲基化反应形成氮杂环丙烷产物.随后63高区域及非对映体选择性地闭环得到五元杂环中间体64, 其中新形成的碳氧键和碳铜键呈反式构象. 64与三氟甲基自由基结合形成高反应活性的三价铜配合物65后进行还原消除得到含氟噁唑啉酮产物61a, 同时再生一价铜配合物催化剂(Scheme 14).
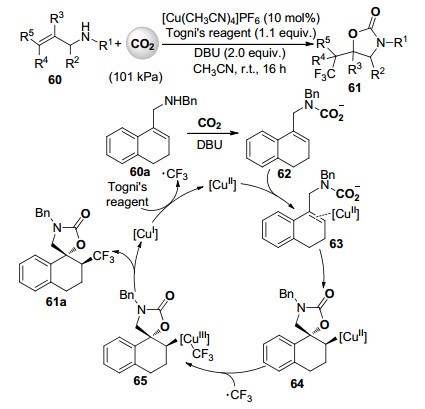 图式14
二氧化碳参与的烯丙胺选择性氧三氟甲基化反应
Scheme14.
Selective oxytrifluoromethylation of allylamines with carbon dioxide
图式14
二氧化碳参与的烯丙胺选择性氧三氟甲基化反应
Scheme14.
Selective oxytrifluoromethylation of allylamines with carbon dioxide
最近, 何良年课题组[44]首次实现了光诱导自由基引发的烯丙胺与二氧化碳的羧化环化反应(Scheme 15).用全氟烷基碘化物为自由基源, 1.5 equiv.的TBD为碱, 一系列烯丙胺化合物66与101 kPa的二氧化碳在可见光照射下于室温条件下进行反应, 高收率, 高化学、区域选择性地合成了一系列全氟烷基官能团化的噁唑啉酮类化合物67.通过一系列的对照及核磁跟踪实验, 作者推测碘全氟烷基化的氨基甲酸盐70是此反应的关键中间体.可能的反应机理见Scheme 15.烯丙胺化合物66与二氧化碳在TBD作用下形成氨基甲酸化合物68.全氟烷基碘化物在光照条件下碳碘键均裂形成全氟烷基自由基加成到68双键上得到自由基物种69, 随后69被之前释放出的碘自由基捕捉后形成碘全氟烷基化的氨基甲酸盐70 (Scheme 15, Path Ⅰ). 69也可与全氟烷基碘化物通过自由基链转移形成70, 同时生成新的全氟烷基自由基进入下一反应循环(Scheme 15, Path Ⅱ).随后70发生分子内亲核取代反应得到全氟烷基取代的噁唑啉酮产物67.此光诱导自由基引发的反应体系也适用于烯丙醇与二氧化碳的羧化环化反应合成全氟烷基取代的环状碳酸酯产物.
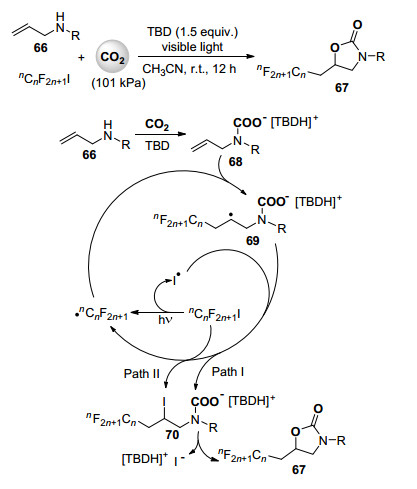 图式15
光诱导自由基引发的烯丙胺与二氧化碳的羧化环化反应
Scheme15.
Photoinduced radical-initiated carboxylative cyclization of allyl amines with carbon dioxide
图式15
光诱导自由基引发的烯丙胺与二氧化碳的羧化环化反应
Scheme15.
Photoinduced radical-initiated carboxylative cyclization of allyl amines with carbon dioxide
2015年, 我们课题组[45]报道了无过渡金属参与的, 三(二甲氨基)膦促进的伯胺, 二氧化碳与α-酮酸酯的多组分羧化反应体系, 所得到产物可方便地进一步闭环得到噁唑啉二酮类化合物71 (Scheme 16), 此两步反应可以通过一锅化串联反应实现.反应过程中三(二甲氨基)膦首先与α-酮酸酯反应得到高亲核反应活性的Kukhtin-Ramirez加合物72, 二氧化碳与伯胺反应得到氨基甲酸盐73[46], 72优先与73进行质子转移生成中间体74, 随后氨基甲酸根亲核进攻释放出膦氧化合物的同时得到氨基甲酸酯化合物75, 其在碱催化下闭环最终得到噁唑啉二酮产物(Scheme 16).由于Kukhtin-Ramirez加合物72的高亲核反应活性[47], 此羧化反应只需在101 kPa的二氧化碳气氛下就可顺利进行.
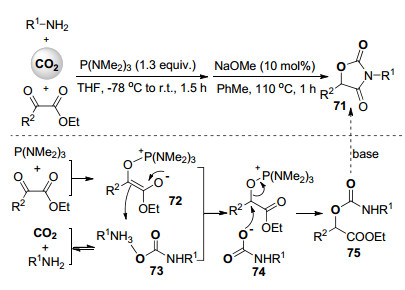 图式16
伯胺、二氧化碳与α-酮酸酯的多组分羧化环化反应
Scheme16.
Multi-component carboxylative cyclization reactions of primary amines, carbon dioxide and α-ketoesters
图式16
伯胺、二氧化碳与α-酮酸酯的多组分羧化环化反应
Scheme16.
Multi-component carboxylative cyclization reactions of primary amines, carbon dioxide and α-ketoesters
6 合成环状碳酸酯
二氧化碳与与环氧烷烃或炔丙醇反应合成环状碳酸酯也是目前二氧化碳转化研究中的一类重要反应[14~18].最近研究者发展了一些二氧化碳参与的环化新反应合成环状碳酸酯.
2012年, Murakami课题组[48]报道了光驱动α-氨基酮76与二氧化碳的羧化环化反应高收率合成包含酰胺取代基的环状碳酸酯77 (Scheme 17).作者首先将α-氨基酮底物76在101 kPa的二氧化碳气氛中置于太阳光下照射8 h后形成高能的氮杂环丁烷化合物78, 此过程中太阳能转化成了化学能.接着向反应体系中加入碳酸铯脱质子形成的79与二氧化碳反应得到包含碳酸根的中间体80, 随后碳酸根中的氧原子亲核进攻氮杂环丁烷环, 以5-exo-tet型的闭环方式形成包含五元环状碳酸酯结构的酰胺基铯盐81, 其质子化后得到产物77 (Scheme 17).
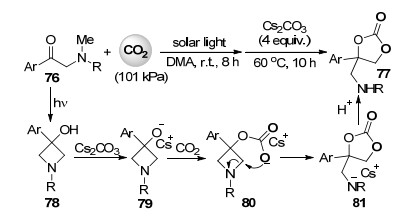 图式17
光驱动α-氨基酮与二氧化碳的羧化环化反应
Scheme17.
Solar-driven carboxylative cyclization reactions of α-amino ketones with carbon dioxide
图式17
光驱动α-氨基酮与二氧化碳的羧化环化反应
Scheme17.
Solar-driven carboxylative cyclization reactions of α-amino ketones with carbon dioxide
通常情况下, 在路易斯酸催化剂和亲核试剂同时存在下, 环氧丙醇/胺82与二氧化碳的环化反应主要通过SN2过程形成环状碳酸酯83.最近, Kleij课题组[49]发现, 在如Scheme 18中所示的铝配合物催化下, 反应体系中无需外加亲核试剂时, 环氧丙醇/胺82与二氧化碳的环化反应主要通过形成中间体84, 然后通过碳酸根中的氧原子分子内亲核进攻(SNi过程)被铝配合物活化的环氧部分, 以5-exo-tet型的环化方式形成五元环状碳酸酯或噁唑啉酮85 (Scheme 18).当在环氧丙胺反应体系中加入亲核性较差的碱如二异丙基乙基胺时, 可促进此SNi反应过程, 从而高选择性地得到噁唑啉酮产物.在此基础上, 作者实现了使用同一种环氧丙醇及环氧丙胺底物通过微调反应条件(如亲核试剂的使用、碱的加入等)控制其反应的化学选择性来合成完全不同结构的两种环化产品.
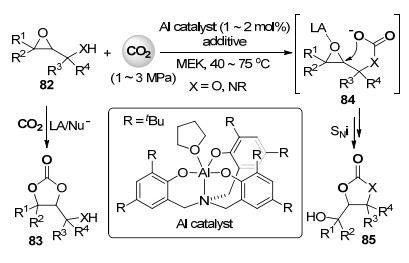 图式18
铝配合物催化环氧丙醇/胺与二氧化碳的环化反应
Scheme18.
Aluminium-catalyzed cyclization reactions of epoxy alcohols and amines with carbon dioxide
图式18
铝配合物催化环氧丙醇/胺与二氧化碳的环化反应
Scheme18.
Aluminium-catalyzed cyclization reactions of epoxy alcohols and amines with carbon dioxide
高烯丙醇及高炔丙醇与二氧化碳的亲电环化反应早在2010年已有报道. 2015年, Johnston课题组[50]报道了在双三氟甲基磺酰亚胺与有机催化剂86 1:1组成的双布朗斯特酸/碱催化剂存在下, 高烯丙醇87与101 kPa的二氧化碳和N-碘代丁二酰亚胺的不对称亲电环化反应, 得到一系列手性的碘代六元环状碳酸酯88 (Scheme 19). N-碘代丁二酰亚胺首先与高烯丙醇中的双键形成碘离子后, 醇羟基与二氧化碳形成的碳酸根在手性有机催化剂的作用下选择性闭环形成产物.作者发现双三氟甲基磺酰亚胺与有机催化剂86形成的氢键网络对反应的转化率的产品的ee值都有关键的促进作用. R基团为间位或对位取代的芳基时, 反应的对映选择性较高, ee值超过90%. R基团为邻位取代的芳基时反应不能进行. R基团为烷基时, 反应的收率和的对映体选择性略有降低.由于产物88中具有高反应活性的碳碘键, 很容易衍生化成多种在精细有机合成中有着重要用途的手性合成子.
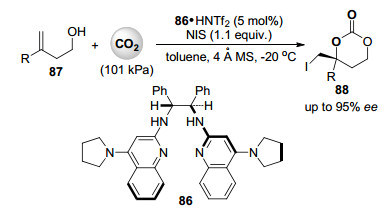 图式19
高烯丙醇与二氧化碳的不对称亲电环化反应
Scheme19.
Enantioselective electrophilic cyclization reactions of homoallylic alcohols and carbon dioxide
图式19
高烯丙醇与二氧化碳的不对称亲电环化反应
Scheme19.
Enantioselective electrophilic cyclization reactions of homoallylic alcohols and carbon dioxide
最近, 成江课题组[51]报道了钯催化炔丙醇, 碘代物和101 kPa二氧化碳的多组分羧化环化反应(Scheme 20), 以高产率合成了一系列环状碳酸酯化合物.此反应中零价钯与碘代物氧化加成后得到的二价钯配合物作为路易斯酸活化炔丙醇89中的碳碳叁键, 通过反式氧钯化实现羧化环化后, 再经还原消除过程在双键上引入芳基、烯基或甲基, 得到最终环状碳酸酯产物90.当使用端炔丙醇底物91时, 只需使用过量的碘苯和添加10 mol%的碘化亚铜, 便可在钯催化下先进行Sonogashira偶联, 然后再进行多组分羧化环化反应得到双键端位上引入两个苯基的噁唑啉酮产物92 (Scheme 20).
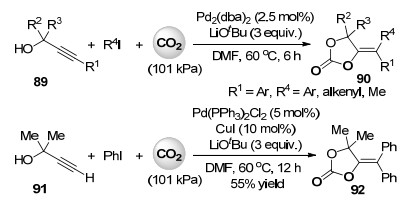 图式20
钯催化炔丙醇、碘代物和二氧化碳的多组分羧化环化反应
Scheme20.
Palladium-catalyzed multi-component carboxylative cyclization reactions of propargyl alcohols, organic iodides and carbon dioxide
图式20
钯催化炔丙醇、碘代物和二氧化碳的多组分羧化环化反应
Scheme20.
Palladium-catalyzed multi-component carboxylative cyclization reactions of propargyl alcohols, organic iodides and carbon dioxide
最近, 侯召民课题组在他们一系列氮杂环卡宾配合物催化的二氧化碳转化研究工作基础上[52, 53], 发展了氮杂环卡宾铜配合物催化的醛与联硼酸频那醇酯, 二氧化碳的硼羧化反应[54], 高收率地合成了一系列可作为锂离子电池电极材料的硼杂环状碳酸酯锂化合物93 (Scheme 21).作者推测的反应机理中, 首先氮杂环卡宾氯化亚铜与叔丁醇锂进行离子交换形成叔丁氧基铜配合物94, 经与联硼酸频那醇酯反应后形成硼烷基铜配合物95, 随后醛插入95的铜硼键形成烷氧基铜中间体96, 二氧化碳插入96中的铜氧键后紧接着氮杂环卡宾铜迁移形成硼杂环状碳酸酯中间体97, 叔丁醇锂与97进行离子交换释放出产物的同时再生叔丁氧基铜配合物94 (Scheme 21).
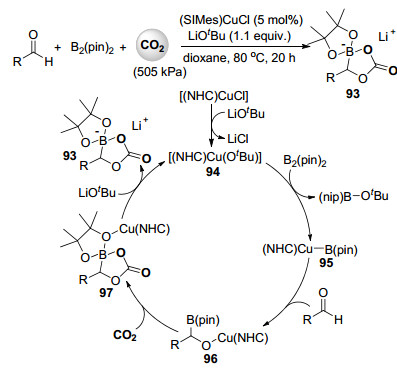 图式21
铜催化醛与联硼酸频那醇酯, 二氧化碳的硼羧化反应
Scheme21.
Copper-catalyzed bora-carboxylation reactions of aldehydes with B2(pin)2 and carbon dioxide
图式21
铜催化醛与联硼酸频那醇酯, 二氧化碳的硼羧化反应
Scheme21.
Copper-catalyzed bora-carboxylation reactions of aldehydes with B2(pin)2 and carbon dioxide
7 合成苯并咪唑及苯并噻唑
二氧化碳加氢还原是目前二氧化碳转化研究中的热点.而在还原剂如硅烷或硼烷等存在下, 二氧化碳可与伯胺或仲胺进行还原性羧化环化反应得到在精细有机合成中有着重要用途的甲胺基或甲酰胺基产物, 此时碳已从二氧化碳的+4价转化成低价态.
2015年, 刘志敏课题组[55]在他们之前二氧化碳转化及离子液体研究工作基础上, 首次发展了室温条件下离子液体催化邻氨基苯硫酚98或邻苯二胺99与三乙氧基硅烷, 及二氧化碳的还原性羧化环化反应, 高收率地得到苯并噻唑(100)及苯并咪唑(101)类化合物(Scheme 22).在所使用的离子液体中, 1-丁基-2, 3-二甲基咪唑乙酸盐([Bmmim][OAc])表现出最好的催化活性, 可重复利用5次而不失活.通过详细的核磁研究, 作者提出了可能的反应机理, 见Scheme 22.二氧化碳首先与[Bmmim][OAc]反应形成类似于氮杂环卡宾二氧化碳加合物的中间体102, 然后102中处于活化状态的二氧化碳插入被[Bmmim][OAc]活化的硅烷中的硅氢键, 形成醛氧基硅烷中间体103.邻氨基苯硫酚或邻苯二胺底物通过与离子液体中的乙酸负离子形成氢键而被活化后再与中间体103反应得到甲酰胺基化合物104, 最后分子内脱水环化得到苯并噻唑或苯并咪唑产物.从机理可以看出, 离子液体对此反应中的邻氨基苯硫酚或邻苯二胺、二氧化碳和硅烷三个反应组分都有活化作用.最近, 李福伟[56]和韩布兴[57]课题组分别发现二甲基亚砜或γ-戊内酯同时作为溶剂和催化剂也可促进此还原性羧化环化反应.
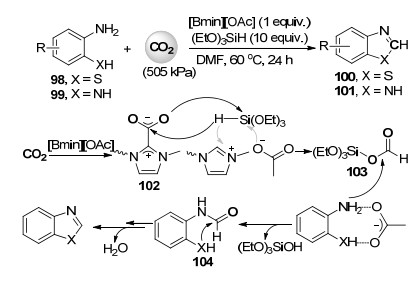 图式22
离子液体催化邻氨基苯硫酚或邻苯二胺与硅烷、二氧化碳的还原性羧化环化反应
Scheme22.
Ionic liquid-catalyzed reductive cyclization of 2-aminothiophenols or o-phenylenediamines with hydrosilane and carbon dioxide
图式22
离子液体催化邻氨基苯硫酚或邻苯二胺与硅烷、二氧化碳的还原性羧化环化反应
Scheme22.
Ionic liquid-catalyzed reductive cyclization of 2-aminothiophenols or o-phenylenediamines with hydrosilane and carbon dioxide
最近, 孙伟课题组[58]报道了全氟苯基硼催化邻苯二胺与苯基硅烷、二氧化碳的还原性羧化环化反应合成苯并咪唑类化合物101 (Scheme 23).机理研究发现此反应体系中主要通过形成受阻路易酸碱对(FLP)来完成催化循环过程.全氟苯基硼、邻苯二胺和二氧化碳首先反应形成FLP和二氧化碳的加合物105, 接着全氟苯基硼活化的苯基硅烷与105反应得到中间体106, 随后全氟苯基硼离去的同时生成的甲酰胺产物经分子内脱水环化得到苯并咪唑产物.
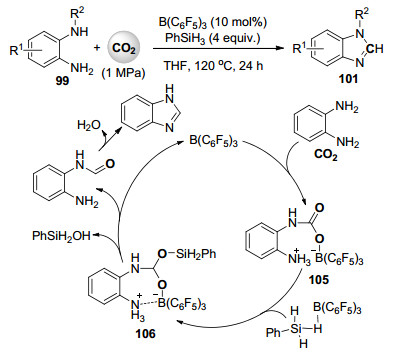 图式23
全氟苯基硼催化邻苯二胺与硅烷、二氧化碳的还原性羧化环化反应
Scheme23.
B(C6F5)3-catalyzed reductive cyclization of o-phen-ylenediamines with hydrosilane and carbon dioxide
图式23
全氟苯基硼催化邻苯二胺与硅烷、二氧化碳的还原性羧化环化反应
Scheme23.
B(C6F5)3-catalyzed reductive cyclization of o-phen-ylenediamines with hydrosilane and carbon dioxide
8 合成环状羧酸化合物
在许多二氧化碳作为反应底物但不参与成环过程环化反应中, 二氧化碳的插入过程可能会促进后续的成环反应, 或者先发生的环化过程产生的高活性的中间体与二氧化碳进行后续反应最终得到环状羧酸产物.
亲电试剂如有机卤代物与二氧化碳的直接还原羧化是目前二氧化碳催化转化中的一类重要新反应[59].最近, Martin课题组[60]报道了镍催化含有β氢的非活化烷基溴代物的还原性环化/二氧化碳羧化串联反应(Scheme 24).使用锰粉作为还原剂, 溴化镍乙二醇二甲醚配合物与邻菲罗啉类配体组成的催化体系能有效抑制β氢消除及二聚等副反应, 在室温及常压条件下便可高效地将一系列含有β氢的非活化烷基溴代物转化成相应环状α, β-不饱和羧酸(Scheme 24).产品的顺反构型可以很容易地通过底物及配体的选择进行控制.当使用伯卤代物及配体L1时, 反应主要得到顺式的产品108; 当使用仲卤代物及配体L2时, 反应主要得到反式的产品109 (Scheme 24).此反应条件温和, 底物适用性宽, 原料易得, 操作简便, 为合成两种构型的环状α, β-不饱和羧酸提供了行之有效的方法.虽然确切的反应机理目前还不够清楚, 但初步的实验表明羧化产物是通过一价镍物种与二氧化碳反应得到的.
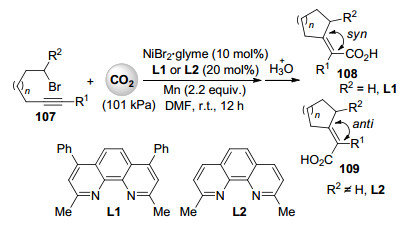 图式24
镍催化非活化烷基溴代物的还原性环化/二氧化碳羧化串联反应
Scheme24.
Ni-catalyzed cascade reductive cyclization/carbox-ylation of unactivated alkyl bromides with carbon dioxide
图式24
镍催化非活化烷基溴代物的还原性环化/二氧化碳羧化串联反应
Scheme24.
Ni-catalyzed cascade reductive cyclization/carbox-ylation of unactivated alkyl bromides with carbon dioxide
2016年, 麻生明课题组[61]报道了镍催化二炔烃110与二乙基锌和101 kPa二氧化碳的氢羧化反应, 在温和条件下高立体及区域选择性地合成了一系列环状共轭2, 4-二烯羧酸111 (Scheme 25).根据详细的对照实验和产物的立体及区域选择性特征, 作者提出的分步反应机理见Scheme 25.首先零价镍优先与反应活性更高的烷基取代的碳碳三键配位形成中间体112.由于芳基取代的碳碳三键的配位, 112与二乙基锌的转金属化反应可高区域选择性地进行, 得到中间体113. β氢消除形成114, 插入芳基取代的碳碳三键得到115, 还原消除后形成的有机锌中间体116与二氧化碳进行羧化反应, 经后处理得到环状共轭2, 4-二烯羧酸产品.
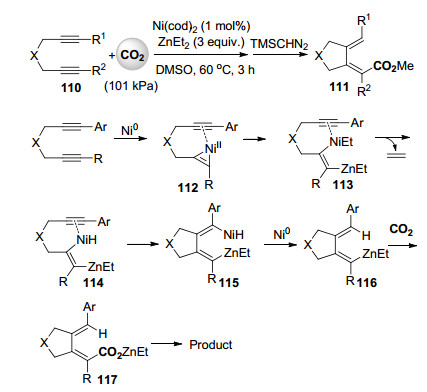 图式25
镍催化二炔烃与二乙基锌、二氧化碳的氢羧化反应
Scheme25.
Nickel-catalyzed three-component hydrocarboxylation of diynes with diethylzinc and carbon dioxide
图式25
镍催化二炔烃与二乙基锌、二氧化碳的氢羧化反应
Scheme25.
Nickel-catalyzed three-component hydrocarboxylation of diynes with diethylzinc and carbon dioxide
最近, 麻生明课题组[62]也报道了邻炔基苯磺酰胺与101 kPa的二氧化碳在3 equiv.的二乙基锌存在下的环化反式氮羧化反应(Scheme 26), 高收率地合成了包含一系列取代基的3-吲哚羧酸化合物.作者提出的可能反应机理包括底物首先与二乙基锌作用发生环化反应形成有机锌中间体121, 其中二乙基锌同时作为碱和活化碳碳叁键的路易斯酸.随后二氧化碳羧化反应形成羧酸锌物种122, 经后处理得到3-吲哚羧酸产品.此反应条件温和, 底物适用性宽, 操作简便.作者以此羧化反应为基础成功合成了已商品化的药物分子Lotronex.
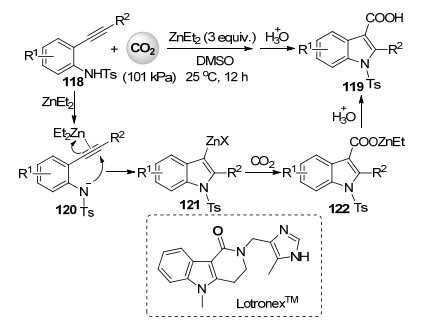 图式26
炔基苯磺酰胺与二氧化碳的环化反式氮羧化反应
Scheme26.
Cyclic anti-azacarboxylation of 2-alkynylanilines with carbon dioxide
图式26
炔基苯磺酰胺与二氧化碳的环化反式氮羧化反应
Scheme26.
Cyclic anti-azacarboxylation of 2-alkynylanilines with carbon dioxide
最近, 我们课题组[63]在之前炔基酮与二氧化碳的羧化环化反应的基础上, 发展了铜催化的邻炔基苯乙酮123与二氧化碳的双羧化反应(Scheme 27), 含有各种官能团的邻炔基苯乙酮与二氧化碳进行羧化/分子内环化/再羧化的串联反应得到一系列异苯并呋喃类二羧酸酯产物124.炔基上的取代基的空间位阻对产物选择性有着重要影响.相比于芳基取代的炔底物, 烷基取代的炔底物显示出更好的双羧化反应选择性.可能的反应机理见Scheme 27.邻炔基苯乙酮首先与一分子的二氧化碳在碱作用下形成β酮酸中间体125, 接着125中的烯醇式氧原子进攻被一价铜活化的碳碳叁键得到包含碳铜键的中间体127, 另外一分子二氧化碳插入后形成的羧基铜物种128与碘甲烷反应得到双羧化的羧酸酯产物.
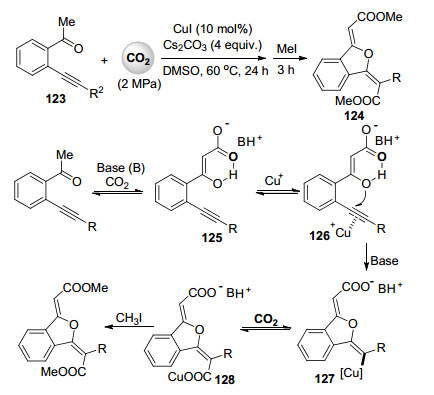 图式27
邻炔基苯乙酮与二氧化碳的双羧化反应
Scheme27.
Double carboxylation of 2-alkynyl acetophenone with carbon dioxide
图式27
邻炔基苯乙酮与二氧化碳的双羧化反应
Scheme27.
Double carboxylation of 2-alkynyl acetophenone with carbon dioxide
9 总结与展望
将二氧化碳作为廉价易得的可再生碳源, 通过环化反应将其转化成高附加值的环状产物的研究有了重要进展.二氧化碳可以和一系列高能试剂反应得到内酰胺、内酯、邻苯二甲酰亚胺、环状酸酐、噁唑啉酮、环状碳酸酯、苯并噻唑及苯并咪唑等重要的杂环化合物.其中有些高效的环化反应由于操作简便, 容易放大, 具有在精细有机合成生产中替代传统方法而实际应用的巨大潜力.但从总体上来说, 由于二氧化碳的反应惰性, 其参与的环化反应类型还不够丰富.大部分环化反应还需在高温高压下或过渡金属催化剂存在下进行.光驱动的二氧化碳参与的环化反应虽有报道但底物适用性不够广泛.因此, 在发展传统的单一依靠化学能转化的二氧化碳环化反应的同时, 通过引入光、电等其他形式的能量, 发展新型的、反应类型更加丰富的、产品结构更加多样且实用的高效二氧化碳环化反应是可以期望的重要突破点.
-
-
[1]
Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365. doi: 10.1021/cr068357u
-
[2]
He, M.; Sun, Y.; Han, B. Angew. Chem., Int. Ed. 2013, 52, 9620. doi: 10.1002/anie.201209384
-
[3]
Aresta, M.; Dibenedetto, A.; Angelini, A. Chem. Rev. 2014, 114, 1709. doi: 10.1021/cr4002758
-
[4]
Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Nat. Commun. 2015, 6, 5933. doi: 10.1038/ncomms6933
-
[5]
何良年, 二氧化碳化学, 科学出版社, 北京, 2013.He, L.-N. Carbon Dioxide Chemistry, Science Publisher, Beijing, 2013.
-
[6]
Huang, K.; Sun, C. L.; Shi, Z.-J. Chem. Soc. Rev. 2011, 40, 2435. doi: 10.1039/c0cs00129e
-
[7]
Tsuji, Y.; Fujihara, T. Chem. Commun. 2012, 48, 9956. doi: 10.1039/c2cc33848c
-
[8]
Yang, Z.-Z.; He, L.-N.; Gao, J.; Liu, A.-H.; Yu, B. Energy Environ. Sci. 2012, 5, 6602. doi: 10.1039/c2ee02774g
-
[9]
Cai, X.; Xie, B. Synthesis 2013, 45, 3305. doi: 10.1055/s-00000084
-
[10]
Wang, S.; Du, G.; Xi, C. Org. Biomol. Chem. 2016, 14, 3666. doi: 10.1039/C6OB00199H
-
[11]
于博, 刘志敏, 科学通报, 2015, 60, 1452.Yu, B.; Liu, Z. M. Chin. Sci. Bull. 2015, 60, 1452.
-
[12]
Yu, B.; He, L.-N. ChemSusChem 2015, 8, 52. doi: 10.1002/cssc.201402837
-
[13]
Rintjema, J.; Kleij, A. W. Synthesis 2016, 48, 3863. doi: 10.1055/s-0035-1562520
-
[14]
Lu, X.-B.; Darensbourg, D. J. Chem. Soc. Rev. 2012, 41, 1462. doi: 10.1039/C1CS15142H
-
[15]
Kielland, N.; Whiteoak, C. J.; Kleij, A. W. Adv. Synth. Catal. 2013, 355, 2115. doi: 10.1002/adsc.201300422
-
[16]
Xu, B.-H.; Wang, J.-Q.; Sun, J.; Huang, Y.; Zhang, J.-P.; Zhang, X.-P.; Zhang, S.-J. Green Chem. 2015, 17, 108. doi: 10.1039/C4GC01754D
-
[17]
Martin, C.; Fiorani, G.; Kleij, A. W. ACS Catal. 2015, 5, 1353. doi: 10.1021/cs5018997
-
[18]
Lang, X.-D.; He, L.-N. Chem. Rec. 2016, 16, 1337. doi: 10.1002/tcr.201500293
-
[19]
Manjolinho, F.; Arndt, M.; Gooßen, K.; Gooßen, L. J. ACS Catal. 2012, 2, 2014. doi: 10.1021/cs300448v
-
[20]
朱庆, 王露, 夏春谷, 刘超, 有机化学, 2016, 36, 2813. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345691.shtmlZhu, Q.; Wang, L.; Xia, C.; Liu, C. Chin. J. Org. Chem. 2016, 36, 2813. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345691.shtml
-
[21]
Zhang, Z.; Liao, L.-L.; Yan, S.-S.; Wang, L.; He, Y.-Q.; Ye, J.-H.; Li, J.; Zhi, Y.-G.; Yu, D.-G. Angew. Chem., Int. Ed. 2016, 55, 7068. doi: 10.1002/anie.201602095
-
[22]
Zhang, Z.; Ju, T.; Ye, J.-H.; Yu, D.-G. Synlett 2017, 28, 741. doi: 10.1055/s-0036-1588403
-
[23]
Wang, S.; Shao, P.; Du, G.; Xi, C. J. Org. Chem. 2016, 81, 6672. doi: 10.1021/acs.joc.6b01318
-
[24]
Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuery, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465. doi: 10.1002/anie.201001035
-
[25]
Sun, S.; Hu, W.-M.; Gu, N.; Cheng, J. Chem. Eur. J. 2016, 22, 18729. doi: 10.1002/chem.v22.52
-
[26]
Sasano, K.; Takaya, J.; Iwasawa, N. J. Am. Chem. Soc. 2013, 135, 10954. doi: 10.1021/ja405503y
-
[27]
Zhang, Z.; Ju, T.; Miao, M.; Han, J.-L.; Zhang, Y.-H.; Zhu, X.-Y.; Ye, J.-H.; Yu, D.-G.; Zhi, Y.-G. Org. Lett. 2017, 19, 396. doi: 10.1021/acs.orglett.6b03601
-
[28]
Xin, Z.; Lescot, C.; Friis, S. D.; Daasbjerg, K.; Skrydstrup, T. Angew. Chem., Int. Ed. 2015, 54, 6862. doi: 10.1002/anie.201500233
-
[29]
Bhunia, A.; Yetra, S. R.; Biju, A. T. Chem. Soc. Rev. 2012, 41, 3140. doi: 10.1039/c2cs15310f
-
[30]
Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. J. Am. Chem. Soc. 2006, 128, 11040. doi: 10.1021/ja064157o
-
[31]
Yoo, W.-J.; Nguyen, T. V. Q.; Kobayashi, S. Angew. Chem., Int. Ed. 2014, 53, 10213. doi: 10.1002/anie.201404692
-
[32]
Zhang, W.-Z.; Li, W.-J.; Zhang, X.; Zhou, H.; Lu, X.-B. Org. Lett. 2010, 12, 4748. doi: 10.1021/ol102172v
-
[33]
Flowers, B. J.; Gautreau-Service, R.; Jessop, P. G. Adv. Synth. Catal. 2008, 350, 2947. doi: 10.1002/adsc.v350:18
-
[34]
Sekine, K.; Ishida, T.; Yamada, T. Angew. Chem., Int. Ed. 2012, 51, 6989. doi: 10.1002/anie.201201399
-
[35]
Sekine, K.; Takayanagi, A.; Kikuchi, S.; Yamada, T. Chem. Commun. 2013, 49, 11320. doi: 10.1039/c3cc47221c
-
[36]
Zhang, W.-Z.; Shi, L.-L.; Liu, C.; Yang, X.-T.; Wang, Y.-B.; Luo, Y.; Lu, X.-B. Org. Chem. Front. 2014, 1, 275. doi: 10.1039/c3qo00047h
-
[37]
Zhang, W.-Z.; Liu, S.; Lu, X.-B. Beilstein J. Org. Chem. 2015, 11, 906. doi: 10.3762/bjoc.11.102
-
[38]
Zhang, W.-Z.; Yang, M.-W.; Lu, X.-B. Green Chem. 2016, 18, 4181. doi: 10.1039/C6GC01346E
-
[39]
Masuda, Y.; Ishida, N.; Murakami, M. J. Am. Chem. Soc. 2015, 137, 14063. doi: 10.1021/jacs.5b10032
-
[40]
Kaicharla, T.; Thangaraj, M.; Biju, A. T. Org. Lett. 2014, 16, 1728. doi: 10.1021/ol500403x
-
[41]
Fujihara, T.; Horimoto, Y.; Mizoe, T.; Sayyed, F. B.; Tani, Y.; Terao, J.; Sakaki, S.; Tsuji, Y. Org. Lett. 2014, 16, 4960. doi: 10.1021/ol502538r
-
[42]
Garcia-Dominguez, P.; Fehr, L.; Rusconi, G.; Nevado, C. Chem. Sci. 2016, 7, 3914 doi: 10.1039/C6SC00419A
-
[43]
Ye, J.-H.; Song, L.; Zhou, W.-J.; Ju, T.; Yin, Z.-B.; Yan, S.-S.; Zhang, Z.; Li, J.; Yu, D.-G. Angew. Chem., Int. Ed. 2016, 55, 10022. doi: 10.1002/anie.201603352
-
[44]
Wang, M.-Y.; Cao, Y.; Liu, X.; Wang, N.; He, L.-N.; Lia, S.-H. Green Chem. 2017, 19, 1240. doi: 10.1039/C6GC03200A
-
[45]
Zhang, W.-Z.; Xia, T.; Yang, X.-T.; Lu, X.-B. Chem. Commun. 2015, 51, 6175. doi: 10.1039/C5CC01530H
-
[46]
Zhang, W.-Z.; Ren, X.; Lu, X.-B. Chin. J. Chem. 2015, 33, 610. doi: 10.1002/cjoc.201500011
-
[47]
Miller, E. J.; Zhao, W.; Herr, J. D.; Radosevich, A. T. Angew. Chem., Int. Ed. 2012, 51, 10605. doi: 10.1002/anie.201205604
-
[48]
Ishida, N.; Shimamoto, Y.; Murakami, M. Angew. Chem., Int. Ed. 2012, 51, 11750. doi: 10.1002/anie.201206166
-
[49]
Rintjema, J.; Epping, R.; Fiorani, G.; Martin, E.; Escudero-Adan, E. C.; Kleij, A. W. Angew. Chem., Int. Ed. 2016, 55, 3972. doi: 10.1002/anie.201511521
-
[50]
Vara, B. A.; Struble, T. J.; Wang, W.; Dobish, M. C.; Johnston, J. N. J. Am. Chem. Soc. 2015, 137, 7302. doi: 10.1021/jacs.5b04425
-
[51]
Sun, S.; Wang, B.; Gu, N.; Yu, J.-T.; Cheng, J. Org. Lett. 2017, 19, 1088. doi: 10.1021/acs.orglett.7b00111
-
[52]
Zhang, L.; Hou, Z. Chem. Sci. 2013, 4, 3395. doi: 10.1039/c3sc51070k
-
[53]
张帅, 李雪冬, 何良年, 化学学报, 2016, 74, 17.Zhang, S.; Li, X.-D.; He, L.-N. Acta Chim. Sinica 2016, 74, 17.
-
[54]
Carry, B.; Zhang, L.; Nishiura, M.; Hou, Z. Angew. Chem., Int. Ed. 2016, 55, 6257. doi: 10.1002/anie.201602278
-
[55]
Gao, X.; Yu, B.; Yang, Z.; Zhao, Y.; Zhang, H.; Hao, L.; Han, B.; Liu, Z. ACS Catal. 2015, 5, 6648. doi: 10.1021/acscatal.5b01874
-
[56]
Lv, H.; Xing, Q.; Yue, C.; Lei, Z.; Li, F. Chem. Commun. 2016, 52, 6545. doi: 10.1039/C6CC01234E
-
[57]
Song, J.; Zhou, B.; Liu, H.; Xie, C.; Meng, Q.; Zhang, Z.; Han, B. Green Chem. 2016, 18, 3956. doi: 10.1039/C6GC01455K
-
[58]
Zhang, Z.; Sun, Q.; Xia, C.; Sun, W. Org. Lett. 2016, 18, 6316. doi: 10.1021/acs.orglett.6b03030
-
[59]
Borjesson, M.; Moragas, T.; Gallego, D.; Martin, R. ACS Catal. 2016, 6, 6739. doi: 10.1021/acscatal.6b02124
-
[60]
Wang, X.; Liu, Y.; Martin, R. J. Am. Chem. Soc. 2015, 137, 6476. doi: 10.1021/jacs.5b03340
-
[61]
Cao, T.; Ma, S. Org. Lett. 2016, 18, 1510. doi: 10.1021/acs.orglett.6b00028
-
[62]
Miao, B.; Li, S.; Li, G.; Ma, S. Org. Lett. 2016, 18, 2556. doi: 10.1021/acs.orglett.6b00884
-
[63]
Zhang, W.-Z.; Yang, M.-W.; Yang, X.-T.; Shi, L.-L.; Wang, H.-B.; Lu, X.-B. Org. Chem. Front. 2016, 3, 217. doi: 10.1039/C5QO00374A
-
[1]
-

图 1 二氧化碳参与的环化新反应合成的杂环化合物
Figure 1 Heterocyclic compounds from new cyclization reactions using carbon dioxide

图式1 二氧化碳参与的邻烯基/芳基苯胺中sp2碳氢键的内酰胺化反应
Scheme 1 Lactamization of sp2-C—H bonds in 2-alkenyl and 2-aryl anilines with carbon dioxide

图式2 无金属参与的邻芳基苯胺中sp2-碳氢键与二氧化碳的内酰胺化反应
Scheme 2 Metal-free lactamization of sp2-C—H bonds in 2-arylanilines with carbon dioxide

图式3 钯催化邻碘苯胺、N-对甲基苯磺酰腙和二氧化碳的多组分反应
Scheme 3 Palladium-catalyzed multi-component reactions of 2-iodoanilines, N-tosylhydrazones and carbon dioxide

图式4 钯催化邻羟基苯乙烯sp2-碳氢键与二氧化碳的内酯化反应
Scheme 4 Palladium-catalyzed lactonization of sp2-C—H bonds in 2-hydroxystyrenes with carbon dioxide

图式5 无过渡金属参与邻杂环芳基苯酚及邻羟基苯乙烯中sp2-碳氢键与二氧化碳的内酯化反应
Scheme 5 Transition-metal-free lactonization of sp2-C—H bonds in 2-arylphenols/ 2-hydroxystyrenes with carbon dioxide

图式6 TBD催化2-炔基吲哚与二氧化碳的羧化环化反应
Scheme 6 TBD-catalyzed carboxylative cyclization reactions of 2-alkynyl indoles with carbon dioxide

图式7 铜催化芳炔、端炔和二氧化碳的多组分羧化环化反应
Scheme 7 Copper-catalyzed multi-component carboxylative cyclization reactions of arynes, terminal alkynes and carbon dioxide

图式8 DBU促进的邻羟基/乙酰胺基苯乙酮与二氧化碳的羧化环化反应
Scheme 8 DBU-promoted carboxylative cyclization reactions of 2-hydroxy and 2-acetamidoacetophenone with carbon dioxide

图式9 丙烯基酮与二氧化碳的羧化环化反应
Scheme 9 Carboxylative cyclization reactions of substituted propenyl ketones with carbon dioxide

图式10 光驱动邻烷基苯基酮与二氧化碳的羧化反应
Scheme 10 Light-driven carboxylation of 2-alkylphenyl ketones with carbon dioxide

图式11 芳炔、异腈和二氧化碳的多组分羧化环化反应
Scheme 11 Multi-component carboxylative cyclization reactions of arynes, isocyanides and carbon dioxide

图式12 镍催化内炔与二氧化碳的双羧化反应合成马来酸酐
Scheme 12 Nickel-catalyzed double carboxylations of internal alkynes with carbon dioxide to synthesize maleic anhydrides

图式13 钯催化炔丙胺, 芳基碘和二氧化碳的多组分羧化环化反应
Scheme 13 Palladium-catalyzed multi-component carboxylative cyclization reactions of propargylamines, aryl iodides and carbon dioxide

图式14 二氧化碳参与的烯丙胺选择性氧三氟甲基化反应
Scheme 14 Selective oxytrifluoromethylation of allylamines with carbon dioxide

图式15 光诱导自由基引发的烯丙胺与二氧化碳的羧化环化反应
Scheme 15 Photoinduced radical-initiated carboxylative cyclization of allyl amines with carbon dioxide

图式16 伯胺、二氧化碳与α-酮酸酯的多组分羧化环化反应
Scheme 16 Multi-component carboxylative cyclization reactions of primary amines, carbon dioxide and α-ketoesters

图式17 光驱动α-氨基酮与二氧化碳的羧化环化反应
Scheme 17 Solar-driven carboxylative cyclization reactions of α-amino ketones with carbon dioxide

图式18 铝配合物催化环氧丙醇/胺与二氧化碳的环化反应
Scheme 18 Aluminium-catalyzed cyclization reactions of epoxy alcohols and amines with carbon dioxide

图式19 高烯丙醇与二氧化碳的不对称亲电环化反应
Scheme 19 Enantioselective electrophilic cyclization reactions of homoallylic alcohols and carbon dioxide

图式20 钯催化炔丙醇、碘代物和二氧化碳的多组分羧化环化反应
Scheme 20 Palladium-catalyzed multi-component carboxylative cyclization reactions of propargyl alcohols, organic iodides and carbon dioxide

图式21 铜催化醛与联硼酸频那醇酯, 二氧化碳的硼羧化反应
Scheme 21 Copper-catalyzed bora-carboxylation reactions of aldehydes with B2(pin)2 and carbon dioxide

图式22 离子液体催化邻氨基苯硫酚或邻苯二胺与硅烷、二氧化碳的还原性羧化环化反应
Scheme 22 Ionic liquid-catalyzed reductive cyclization of 2-aminothiophenols or o-phenylenediamines with hydrosilane and carbon dioxide

图式23 全氟苯基硼催化邻苯二胺与硅烷、二氧化碳的还原性羧化环化反应
Scheme 23 B(C6F5)3-catalyzed reductive cyclization of o-phen-ylenediamines with hydrosilane and carbon dioxide

图式24 镍催化非活化烷基溴代物的还原性环化/二氧化碳羧化串联反应
Scheme 24 Ni-catalyzed cascade reductive cyclization/carbox-ylation of unactivated alkyl bromides with carbon dioxide

图式25 镍催化二炔烃与二乙基锌、二氧化碳的氢羧化反应
Scheme 25 Nickel-catalyzed three-component hydrocarboxylation of diynes with diethylzinc and carbon dioxide

图式26 炔基苯磺酰胺与二氧化碳的环化反式氮羧化反应
Scheme 26 Cyclic anti-azacarboxylation of 2-alkynylanilines with carbon dioxide
-


 下载:
下载:



























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 121
- 文章访问数: 6346
- HTML全文浏览量: 1693
 下载:
下载:
