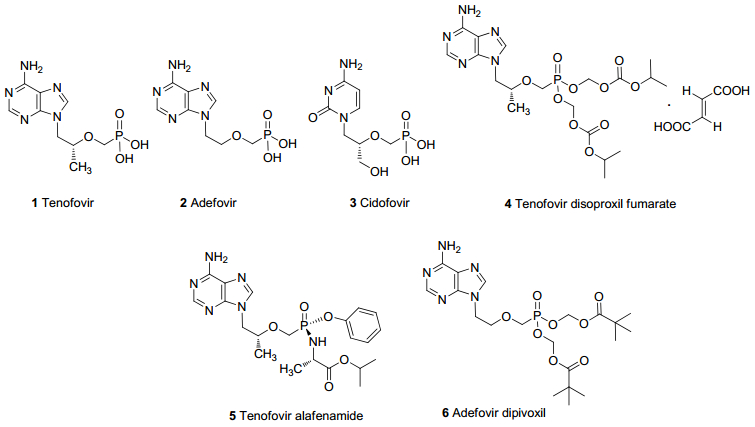
图 1.
第一代ANPs代表药物及其已上市前药
Figure 1.
The first generation ANPs drugs and their marketed prodrugs

核苷类抗病毒药物是临床上广泛应用的一类治疗艾滋病毒(Human immunodeficiency virus, HIV)、乙肝病毒(Hepatitis B virus, HBV)、疱疹病毒(Herpes virus, HSV)等感染的药物[1].核苷类药物作为病毒聚合酶或逆转录酶的抑制剂, 在进入细胞后, 需进行逐步磷酸化转变为三磷酸核苷类似物发挥作用, 但其在体内的磷酸化尤其是第一步磷酸化是非常困难的, 这极大地限制了药物抗病毒活性的发挥, 因此膦酸核苷类抗病毒药应运而生[2].膦酸核苷类抗病毒药按侧链结构主要分为两大类, 分别为环状膦酸核苷(Cyclic nucleoside phosphonates, CNPs)和开环膦酸核苷(Acyclic nucleoside phosphonates, ANPs). CNPs是真正意义上的核苷酸类似物, 含有碱基、糖环和磷酸, 但结构中的糖苷键, 磷酸酯键使其生物稳定性不佳, 进一步磷酸化较困难, 药效不高, 此类药物成功的例子较少[3].相反, ANPs结构中没有糖苷键, 在侧链氧原子和磷酸基团中间插入了亚甲基桥, 稳定性大幅提高; 此外开环侧链柔性更强, 利于药物与不同病毒酶的活性位点相互作用, 活性也得到改善[4].第一代ANPs的代表药物, 替诺福韦、阿德福韦和西多福韦(化合物1~3, 图 1), 具有非常好的抗DNA病毒和逆转录病毒感染作用[5].遗憾的是, 此类药物结构中膦酸盐游离电荷的影响使得药物的生物利用度和细胞穿透性较低; 此外作为人体阴离子转运体的底物, 它们会被动聚集在肾小管, 使肾脏中的药物浓度高于其他组织, 从而引起严重的肾毒性, 使得此类药物的临床应用受到极大的限制[6].在第一代ANPs结构基础上, 对膦酸基团进行保护, 将其制成膦酸酯或膦酰胺前药, 药物的生物利用度大幅提高, 毒副作用也得到了极大改善, 前药研究策略是此类药物的研究方向[7], 富马酸替诺福韦二吡呋酯、替诺福韦艾拉酚胺和阿德福韦酯(化合物4~6, 图 1)的成功上市也证明了这一研究的可行性.此前, 杨康辉(2008年)、王世潇(2010年)、李文保(2012年)[8]等分别以不同的前药形式对核苷类抗病毒药物进行了总结.近几年来, 核苷类抗病毒前药的研究越来越热, 尤其是2016年替诺福韦艾拉酚胺的成功上市, 证明了磷酸酯类前药具有重要应用前景, 也越来越受到研究者们重视.因此, 本文以第一代ANPs结构出发, 对开环膦酸核苷类前药的研究和近几年的最新进展进行综述.
替诺福韦(1, Tenofovir, TFV, 图 1)是由Gilead开发的开环膦酸核苷类化合物, 具有良好的抗HBV和抗HIV活性, 但它的口服生物利用率较低, 严重影响了药效的发挥.
富马酸替诺福韦二吡呋酯(4, Tenofovir disoproxil fumarate, TDF, 图 1)是替诺福韦的酯化前药, 具有较高的水溶性, 在体内吸收后被降解成为替诺福韦, 进而转变为活性代谢产物替诺福韦双磷酸盐, 发挥抗病毒作用, 替诺福韦的双磷酸盐的半衰期较长, 因此药效比较持久[9]. TDF于2001年被美国食品药品监督管理局(FDA)批准用于HIV的治疗, 2008年被批准用于HBV的治疗[10].与TFV相比, TDF具有更高的口服生物利用度和被细胞摄取能力, 它的二磷酸盐在细胞内的浓度能是TFV的1000倍. TDF半衰期约为72 h, 每日给药一次即可, 大大提高了患者服药的依从性, 且抗病毒活性好, 耐药率低, 目前已成为治疗HBV和HIV的一线药物[11].
替诺福韦艾拉酚胺(5, Tenofovir alafenamide, TAF, 图 1)是一种TFV的膦酰胺前体药物[12], 于2016年被FDA批准上市用于治疗成人慢性乙型肝炎, 是近10年来FDA批准的唯一一个用于治疗慢性乙肝的新药.基于TAF的复方制剂也被批准用于治疗HIV感染. TAF口服吸收迅速, 0.48 h即可达到血药浓度的峰值.其亲脂性强, 可通过被动扩散和转运体摄取进入肝细胞, 随后经酶水解、磷酸化形成二磷酸替诺福韦发挥活性[13].与TDF相比, TAF有更好的血浆稳定性, 更强的细胞穿透力, 能直接穿透细胞膜到达淋巴细胞. TAF半衰期比TDF长, 只需要少于TDF十分之一的剂量就可以达到同样的抗病毒疗效, 因此大大降低了代谢物TFV暴露于血浆引发的毒性风险.同时靶向性好, 其长期使用产生的耐药性也会更低, 应用前景优于TDF[14].
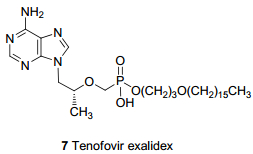
除已上市的TFV前药外, 研究者们还设计了许多其他结构类型的前药.如Tenofovir exalidex (7, TXL, 即CMX157, 图 2), 是由ContraVir公司开发出的一种含长脂肪链的TFV膦酯型肝靶向前药[15], 其独特的结构使其可以利用天然脂质摄取途径将药物送至肝脏, 以减少TFV在其他组织的药物浓度, 从而降低肾脏不良反应[16].在健康志愿者中进行的Ib期临床研究显示[17], TXL具有优良的安全性、耐受性和药物分布.在慢性HBV感染的患者中进行的IIa期多重递增剂量临床试验也已完成[18], 结果显示接受TXL治疗的患者HBV病毒载量平均降低高达99%, 并且达到与标准治疗方案相当的抗病毒活性时, 其剂量仅为标准方案剂量的1/12.此外, 研究还发现与服用标准治疗方案的患者相比, TXL似乎不容易在血液中分解成的TFV.以上结果表明TXL有潜力成为活性高、剂量低、副作用小、安全性高和作用机制独特的肝靶向抗HBV药物.如果后期的临床试验结果良好, TXL将成为HBV联合治疗方案中最安全高效的成份, 为慢性乙肝感染者带来福音.
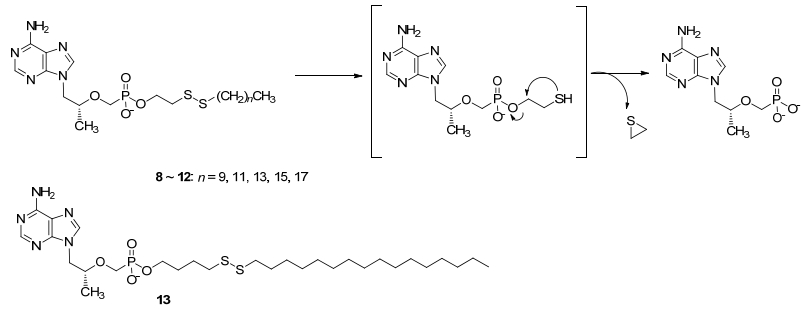
除TXL外, Giesler等[19]还报道了一类含二硫键的脂质化TFV前药8~12(图 3).此类前药具有非常好的抗HIV-1活性, EC50值仅为0.6~8.5 nmol/L; 对HBV也具有较好抑制活性, EC50值为20 nmol/L.它们在血浆中非常稳定, 半衰期超过24 h.其治疗指数高于TFV, 但低于TDF.这可能与前药被还原形成β-巯基乙醇, 并进一步代谢生成硫代环丙烷产生毒性有关(图 3).因此, Giesler等[20]对结构进行优化, 将连接链延长合成了二代含二硫键的脂质化TFV前药, 其中化合物13(图 3)治疗指数>100000, 对HIV-1的抑制活性EC50值仅为0.5 nmol/L, 具有进一步开发价值.
阿德福韦(2, Adefovir, 图 1)是开环腺嘌呤膦酸核苷.阿德福韦在体内和体外都有广泛的抗病毒活性, 但在肠道通透性较差, 膦酸基团的强极性导致口服生物利用度很低.药代动力学研究表明, 在静脉注射后的24 h内, 超过98%的未经过修饰的阿德福韦分子被尿液排出, 在体内无法达到血药浓度的要求.
阿德福韦酯(6, Adefovir dipivoxil, ADV, 图 1)是阿德福韦的酯化前药, 2002年被FDA批准用于治疗慢性乙型肝炎感染, 是FDA批准的第1个治疗慢性乙肝的新药, 商品名贺维力.口服后在体内水解为阿德福韦发挥抗病毒作用[21]. ADV通过人肠上皮细胞的能力比阿德福韦高10倍, 口服生物利用度可提高到20%. ADV吸收后约45%迅速代谢为阿德福韦, 经肾小球过滤和肾小管主动分泌的方式排出体外. ADV的耐药发生率低, 发生时间较晚, 长期用药安全性较高, 适用于长期的抗病毒治疗[22]. ADV对嗜肝病毒、逆转录病毒及痤疮病毒都具有明显的抑制作用.除用于治疗慢性乙肝外, 晚期艾滋病患者使用也能延长存活时间[23].
帕拉德福韦(14, Pradefovir, PDV, 图 4)是由西安新通公司与美国Ligand公司合作利用HepDirect技术设计合成的阿德福韦肝靶向膦酸酯前药[24]. HepDirect技术采用化学修饰的方法, 将药物分子制成无生物活性的前药.此类前药结构中包含一个4-芳基环状取代基, 这一特殊结构使得其对由细胞色素P450酶催化的氧化断裂反应敏感, 可以特异性地被CYP3A4氧化, 释放活性药物分子.因CYP3A4主要是在肝脏实质细胞中表达, HepDirect前药可以特异性靶向到肝实质细胞中, 因此肝部的药物浓度较高, 而其他组织药物浓度较低, 因阿德福韦引起的剂量限制性的肾毒性也更小[25]. PDV口服后吸收迅速, Tmax约为1.0~1.2 h, 吸收后能够迅速转化为阿德福韦, 半衰期约为10 h.与ADV相比, PDV抗病毒效率更高, 抗耐药性更强, 同时解决了ADV毒性大的问题[26].
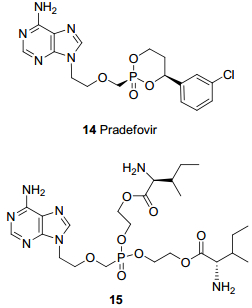
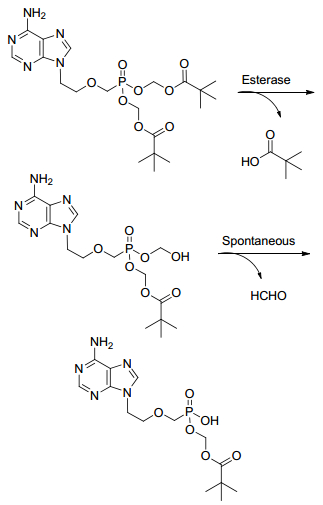
阿德福韦膦酸双L-异亮氨酸乙酯(15, 图 4)[27]是阿德福韦氨基酸酯前药中具有代表性的一个化合物, 其体外抗HBV活性是ADV的五倍(EC50为0.95 μmol/L), 选择性指数大于69000, 分别为ADV和拉米夫定的60倍和24倍.体外稳定性高于ADV, 半衰期约4.5 h.化合物15与ADV相比结构中最大的不同就是, 连接膦酰基与氨基酸的是二碳链, 且侧链为氨基酸酯, 因此不会如ADV的代谢那样生成甲醛和新戊酸, 带来潜在的毒性(图 5)[4].
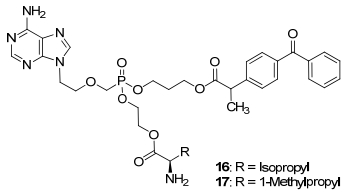
ADV吸收后迅速代谢为阿德福韦, 经肾小球过滤和肾小管主动分泌的方式排出体外, ADV与阿德福韦相比肾毒性已大幅降低, 以10 mg/d的剂量长期应用安全性良好, 但当剂量大于30 mg/d时, 肾毒性发生率为22%~50%.不良反应的发生与人有机阴离子转运蛋白-1 (hOAT-1)对药物的聚集作用有关. hOAT-1对阿德福韦酯有较强的亲和力, 可主动摄取阿德福韦酯, 使其在肾脏近曲小管周围有较高的药物浓度[28].有研究表明, 非甾体抗炎药如布洛芬、酮洛芬等可通过抑制hOAT-1的活性降低阿德福韦的肾毒性.因此Fu等[29]将非甾体抗炎药和L-氨基酸引入阿德福韦结构中制成了混合膦酸酯类化合物16和17(图 6), 体外药理活性研究表明, 16和17活性高于ADV, IC50分别为0.51和0.73μmol/L, 选择性指数分别为1697.64和881.92, 比ADV的毒性更低, 且稳定性更好.
西多福韦(3, Cidofovir, HPMPC, 图 1)是美国Gilead公司开发的胞嘧啶开环核苷膦酸类抗病毒药. 1996年FDA批准其用于治疗艾滋病患者巨细胞病毒(CMV)性视网膜炎.西多福韦具有强大的抗CMV活性, 疗效优于更昔洛韦, 并且对某些耐更昔洛韦或膦甲酸的病毒株也有活性.与其它抗CMV药物相比, 西多福韦疗效更加持久[30].
西多福韦的口服生物利用度低, 小于5.3%, 24 h内90%以原形尿的形式排出.西多福韦最常见的不良反应是剂量限制性肾毒性, 发生率达59%, 严重时会造成不可逆的肾毒性.西多福韦还存在中性粒细胞减少、代谢性酸中毒和眼内压降低等不良反应.西多福韦是人体阴离子转运体的底物, 阴离子的转运体将西多福韦聚集在肾小管, 使肾脏中的药物浓度高于其他组织而引起药物肾毒性.局部使用或病灶内注射药品最常见的不良反应是局部炎症, 严重时会导致患者中断给药[31].
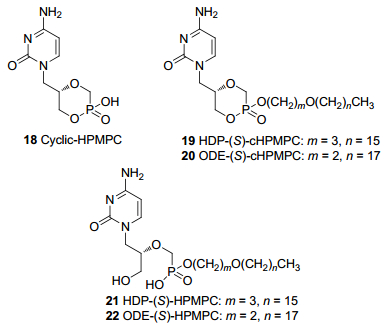
环-西多福韦(18, 图 7)是由西多福韦环合形成的内酯型前药, 化学性质稳定.在细胞内, 能被磷酯酶催化转变成西多福韦.与西多福韦相比口服生物利用度和肾毒性均有改善[32].西多福韦和环-西多福韦均通过细胞的胞饮作用缓慢吸收, 因此, 它们的口服生物利用度有限.以西多福韦和环-西多福韦为母核, 设计一系列的长链脂肪基前药[33], 如十六烷丙氧基-环-西多福韦(19)、十八烷乙氧基-环-西多福韦(20)、十六烷丙氧基-西多福韦(21)、十八烷乙氧基-西多福韦(22) (图 7)等, 可以掩蔽膦酸基团, 明显提高药物的稳定性和脂溶性, 从而提高药物的口服生物利用度, 降低毒副作用.
其中Brincidofovir (21, CMX001, 图 7)是美国Chimerix生物制药公司开发的西多福韦的十六烷丙氧基脂质化前体药物, 在血浆中保持原型, 但进入靶细胞后, 可在细胞内产生高水平的西多福韦及其活性形式西多福韦二磷酸酯, 抑制DNA聚合酶, 导致病毒DNA链延长终止, 从而发挥强大的抗病毒活性[15, 34]. Brincidofovir可在体内缓慢释放西多福韦, 使抗病毒的作用时间显著延长. Brincidofovir与西多福韦结构和药理机制相似, 但肾毒性比西多福韦小, 可口服, 生物利用度更高.遗憾的是, Brincidofovir在治疗CMV感染的三期临床试验研究中没有达到一级临床终点, 临床试验被迫中止[35].目前Chimerix正继续开展此药用于腺病毒和天花治疗的临床试验[36].该化合物还被用于测试埃博拉病毒的治疗[37].
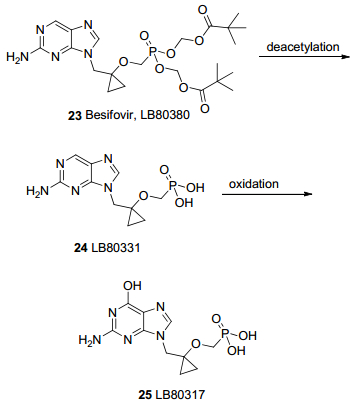
Besifovir (23, LB80380)是一种新型鸟嘌呤核苷膦酸酯型前药, 口服后在肝脏和肠道中转化为LB80331 (24), 然后进一步代谢为化合物LB80317 (25)发挥抗病毒活性(图 8). LB80317是该药物的活性形式, 它是单膦酸鸟苷的类似物, 作用与替诺福韦和阿德福韦类似, 同样因游离膦酸基团影响, 生物利用度低, 毒副作用大, 需要制成前药即Besifovir应用[38]. Besifovir对HBV野生病毒株和拉米夫定耐药株均有良好抑制活性. Besifovir口服后迅速吸收, 且随着剂量增大, 血药浓度呈线性增加.人体中, 口服剂量超过60 mg即对HBV有较好抑制作用.以60 mg剂量为例, 其血药浓度最高为397 ng/mL, 2 h达峰, 半衰期为3 h; 代谢物及原药主要通过肾脏排泄[39]. Besifovir安全性较好, 没有明显的肾毒性和骨毒性[40].一项在亚洲慢性乙肝患者人群中进行的, 对比Besifovir与恩替卡韦安全性和有效性的多中心随机IIb期临床试验表明[41], 在48周的治疗过程中, 90和150 mg/d两个剂量的Besifovir治疗初治慢乙肝患者的疗效不次于0.5 mg/d的恩替卡韦. Besifovir唯一明显的副作用就是血浆中左旋肉碱水平的降低, 需要及时补充肉碱.随后, 研究者们将治疗时间延长至96周, 比较Besifovir 90和150 mg/d与恩替卡韦0.5 mg/d的长期治疗效果差异.结果表明[42], 三组患者在病毒下降、谷丙转氨酶复常、HBeAg转阴方面无明显差异, 三组均无患者出现耐药.可见, Besifovir对慢性乙肝患者的长期抗病毒疗效与恩替卡韦无明显差异, 且安全性和耐受性良好[43].
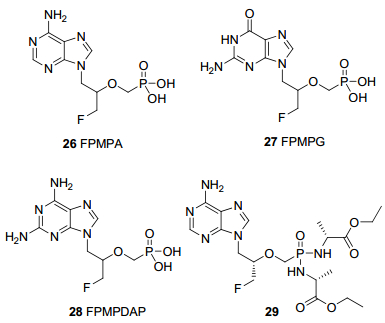
为了获得更多更有效的ANPs, 研究者们设计并合成了许多开环侧链氟取代的类似物(FPMP), 如3-氟- 2-(磷酸甲氧基)丙基取代的腺嘌呤(26)、鸟嘌呤(27)和2, 6-二氨基嘌呤(28), 即FPMPA、FPMPG和FPMPDAP, 图 9)等[44].
与3-羟基-2-(磷酸甲氧基)丙基取代的嘌呤类似物(HPMP)不同, FPMP对多种逆转录病毒有较好选择性和抑制活性, 而对多数DNA病毒活性不佳[45].此外, FPMPA和FPMPDAP两种异构体活性差异很大, (S)-FPMPA和(R)-FPMPDAP对HIV-1和HIV-2的抑制活性EC50分别为8和4.3 μmol/L, 而它们的对映异构体几乎无活性.鸟嘌呤类似物FPMPG两种异构体活性相似, 对HIV-1和HIV-2的抑制活性EC50均为5 μmol/L.将(S)-FPMPA的膦酸基团用丙氨酸掩蔽起来可得到双磷酰胺前药29(图 9), 29具有比原药更强的细胞渗透性, 抗HIV活性提高了十几倍, EC50为0.54 μmol/L.
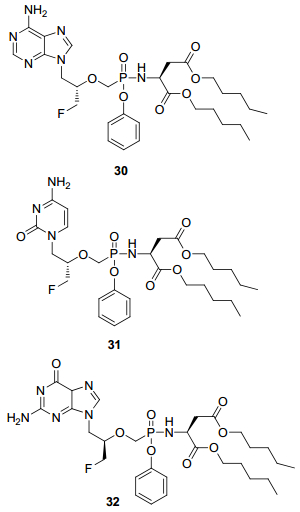
最近, Min Luo等[45]参考已上市的替诺福韦艾拉酚胺, 将不同碱基的FPMP制成天冬氨酸戊酯-膦酰胺型前药30~32(图 10), 不仅使FPMPs的抗病毒活性大幅提高, 还将抗病毒谱扩展到乙肝病毒和疱疹病毒等DNA病毒.其中活性最好的化合物30在TZM-bl细胞抑制HIV的活性的EC50为4 nmol/L, 在PBMCs细胞EC50仅为3 nmol/L; 此外对HBV和VZV也有较好活性, EC50分别为10和50 nmol/L.化合物30在人血浆中稳定, 在肝微粒体细胞中可迅速代谢为原药, 半衰期仅为2 min.
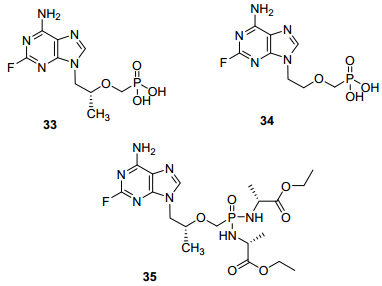
与阿德福韦和替诺福韦相比, 碱基2位引入氟原子形成的化合物33和34(图 11), 对RNA病毒的活性完全丧失, 对DNA病毒活性大幅降低[46].将33制成双磷酰胺前药35(图 11)后, 对HIV有中等抑制活性(EC50为5.5 μmol/L), 这可能与其生物利用度提高有关[47].由此可见, 对ANPs的嘌呤碱基2位氟取代改造于其抗病毒活性而言并无效果.
开环膦酸核苷类抗病毒药, 在临床上病毒感染的治疗中起到了极大的作用.但因其结构中膦酸盐游离电荷的影响使得药物的生物利用度较低, 毒性较大.膦酸酯前药替诺福韦二吡呋酯和阿德福韦酯的广泛应用为此类药物的发展和推广起到了积极作用.但二者的长期毒性同样不可忽视, 这与其不够优化的前药形式直接相关.替诺福韦艾拉酚胺作为替诺福韦膦酰胺前体药物, 不会代谢产生甲醛和新戊酸等毒性代谢物, 应用前景良好. HepDirect肝靶向技术的应用给药物创新带来了新的契机, 利用HepDirect技术设计合成的阿德福韦肝靶向膦酸酯前药帕拉德福韦使阿德福韦获得了新的活力.由西安新通申报的肝靶向1.1类化学新药甲磺酸帕拉德福韦目前临床研究进展良好, 预计可在近几年上市.与嘌呤核苷类ANPs相比, 嘧啶核苷类的西多福韦前药研究成果较少, 目前还没有其前药上市, Brincidofovir抗CMV Ⅲ期临床试验的失败更为西多福韦前药的研究带来阴影.但Brincidofovir临床试验的失败可能与其不当的前药形式有关, 选择制备其他的生物前体药物, 改变前药活化机制, 有望得到适用于临床的广谱抗病毒药物. ANPs前药的研究是一项迫切又任重道远的工程, 通过广大研究者的共同努力, 相信在未来的几年里, 必能获得突破性的研究进展, 为病毒感染的治疗提供更好的选择.
(a) De Clercq, E. Biochem. Pharmacol. 2013, 86, 711.
(b) De Clercq, E. Adv. Pharmacol. 2013, 67, 293.
(c) De Clercq, E.; Holy, A. Nat. Rev. Drug Discovery 2005, 4, 928.
(d) Liu, K.; Xie, L. Acta Pharm. Sin. 2006, 41, 689 (in Chinese).
(刘琨, 谢蓝, 药学学报, 2006, 41, 689.
(a) De Clercq, E. Expert. Rev. Anti-Infect. Ther. 2003, 1, 21.
(b) De Clercq, E. Biochem. Pharmacol. 2011, 82, 99. (c) De Clercq, E. Med. Res. Rev. 2013, 33, 1278.
Peng, Y. M.; Yu, W. Q.; Li, E. T.; Kang, J. F.; Wang, Y. F.; Yang, Q. H.; Liu, B. J.; Zhang, J. M.; Li, L. Y.; Wu, J.; Jiang, J. H.; Wang, Q. D.; Chang, J. B. J. Med. Chem. 2016, 59, 3661. doi: 10.1021/acs.jmedchem.5b01807
Pertusati, F.; Serpi, M.; McGuigan, C. Antivir. Chem. Chemother. 2012, 22, 181. doi: 10.3851/IMP2012
De Clercq, E. Clin. Microbiol. Rev. 2003, 16, 569. doi: 10.1128/CMR.16.4.569-596.2003
De Clercq, E. Antiviral Res. 2007, 75, 1. doi: 10.1016/j.antiviral.2006.10.006
(a) Krecmerova, M. Mini-Rev. Med. Chem. 2017, 17, 818.
(b) Hecker, S. J.; Erion, M. D. J. Med. Chem. 2008, 51, 2328.
(c) Wiemer, A. J.; Wiemer, D. F. Top. Curr. Chem. 2015, 360, 115.
(d) Xie, M. S.; Chen, Y. G.; Wu, X. X.; Qu, G. R.; Guo, H. M. Org. Lett. 2018, 20, 1212.
(a) Yang, K. H.; Wang, J. T.; Li, Q. B.; Xu, W. F. Food Drug 2018, 10, 60 (in Chinese).
(杨康辉, 王举涛, 李乾斌, 徐文方, 食品与药品, 2018, 10, 60.)
(b) Wang, S. X.; Duan, H. D.; Meng, X.; Qin, D. W.; Wang, L. Z.; Li, X. M. J. Beijing Union Univ. (Nat. Sci.) 2010, 24, 29 (in Chinese).
(王世潇, 段洪东, 孟霞, 秦大伟, 王利振, 李晓萌, 北京联合大学学报(自然科学版), 2010, 24, 29.)
(c) Li, W. B.; Dong, F. H.; Sun, C. J. Prog. Pharm. Sci. 2012, 36, 300 (in Chinese).
(李文保, 董芳华, 孙昌俊, 药学进展, 2012, 36, 300.
Kearney, B. P.; Flaherty, J. F.; Shah, J. Clin. Pharmacokinet. 2004, 43, 595. doi: 10.2165/00003088-200443090-00003
Nelson, M. R.; Katlama, C.; Montaner, J. S.; Cooper, D. A.; Gazzard, B.; Clotet, B.; Lazzari, A.; Schewe, K.; Lange, J.; Wyatt, C. AIDS 2007, 21, 1273. doi: 10.1097/QAD.0b013e3280b07b33
Barditch-Crovo, P.; Deeks, S. G.; Collier, A.; Safrin, S.; Coakley, D. F.; Miller, M.; Kearney, B. P.; Coleman, R. L.; Lamy, P. D.; Kahn, J. O. Antimicrob. Agents Chemother. 2001, 45, 2733. doi: 10.1128/AAC.45.10.2733-2739.2001
Ray, A. S.; Fordyce, M. W.; Hitchcock, M. J. M. Antiviral Res. 2016, 125, 63. doi: 10.1016/j.antiviral.2015.11.009
Ruane, P. J.; DeJesus, E.; Berger, D.; Markowitz, M.; Bredeek, U. F.; Callebaut, C.; Zhong, L.; Ramanathan, S.; Rhee, M. S.; Fordyce, M. W. J. Acquired Immune Defic. Syndr. 2013, 63, 449. doi: 10.1097/QAI.0b013e3182965d45
(a) Sax, P. E.; Zolopa, A.; Brar, I.; Elion, R.; Ortiz, R.; Post, F.; Wang, H.; Callebaut, C.; Martin, H.; Fordyce, M. W. J. J. Acquired Immune Defic. Syndr. 2014, 67, 52.
(b) Zhang, X. Q. Acta. Pharm. Sin. 2015, 50, 509 (in Chinese).
(张兴权, 药学学报, 2015, 50, 509.
Painter, G. R.; Almond, M. R.; Trost, L. C.; Lampert, B. M.; Neyts, J.; De Clercq, E.; Korba, B. E.; Aldern, K. A.; Beadle, J. R.; Hostetler, K. Y. Antimicrob. Agents Chemother. 2007, 51, 3505. doi: 10.1128/AAC.00460-07
Lanier, E. R.; Lampert, B.; Trost, L.; Painter, G.; Almond, M. Antiviral Ther. 2008, 13, A6. http://www.ncbi.nlm.nih.gov/pubmed/21499452
Tanwandee, T.; Chatsiricharoenkul, S.; Thongsawat, S.; Sukeepaisarnjaroen, W.; Tangkijvanich, P.; Komolmit, P.; Avihingsanon, A.; Piratvisuth, T.; Sunthi, P.; Mahasanprasert, T. J. Hepatol. 2017, 66, S24. http://www.ncbi.nlm.nih.gov/pubmed/26339796
Chatsiricharoekul, S.; Jutasompakorn, P.; Niyomnaitham, S.; Matkovits, T.; Conover, M.; Cobb, J.; Greytok, J.; Sullivan-Bolyai, J. Clin. Pharmacol. Ther. 2017, 101, S48. http://www.ncbi.nlm.nih.gov/pubmed/25880550
Giesler, K. E.; Marengo, J.; Liotta, D. C. J. Med. Chem. 2016, 59, 7097. doi: 10.1021/acs.jmedchem.6b00428
Giesler, K. E.; Liotta, D. C. J. Med. Chem. 2016, 59, 10244. doi: 10.1021/acs.jmedchem.6b01292
Marcellin, P.; Chang, T.-T.; Lim, S. G.; Tong, M. J.; Sievert, W.; Shiffman, M. L.; Jeffers, L.; Goodman, Z.; Wulfsohn, M. S.; Xiong, S. N. Engl. J. Med. 2003, 348, 808. doi: 10.1056/NEJMoa020681
Marcellin, P.; Chang, T.-T.; Lim, S. G. L.; Sievert, W.; Tong, M.; Arterburn, S.; Borroto-Esoda, K.; Frederick, D.; Rousseau, F. Hepatology 2008, 48, 750. doi: 10.1002/hep.v48:3
Ingiliz, P.; Valantin, M.-A.; Thibault, V.; Duvivier, C.; Dominguez, S.; Katlama, C.; Poynard, T.; Benhamou, Y. Antivir. Ther. 2008, 13, 895.
Reddy, K. R.; Matelich, M. C.; Ugarkar, B. G.; Gomez-Galeno, J. E.; DaRe, J.; Ollis, K.; Sun, Z.; Craigo, W.; Colby, T. J.; Fujitaki, J. M. J. Med. Chem. 2008, 51, 666. doi: 10.1021/jm7012216
Lin, C. C.; Fang, C.; Benetton, S.; Xu, G. F.; Yeh, U. T. Antimicrob. Agents Chemother. 2006, 50, 2926. doi: 10.1128/AAC.01566-05
Ding, Y.; Zhang, H.; Li, X.; Li, C.; Chen, G.; Chen, H.; Wu, M.; Niu, J. Hepatol. Int. 2017, 11, 390. doi: 10.1007/s12072-017-9797-y
Fu, X.; Jiang, S.; Li, C.; Xin, J.; Yang, Y.; Ji, R. Bioorg. Med. Chem. Lett. 2007, 17, 465. doi: 10.1016/j.bmcl.2006.10.021
傅晓钟, 王永林, 兰燕宇, 王爱民, 欧瑜, 罗春, 李燕, 药学学报, 2010, 45, 1017. http://www.cnki.com.cn/Article/CJFDTotal-YXXB201008017.htmFu, X. Z.; Wang, Y. L.; Lan, Y. Y.; Wang, A. M.; Ou, Y.; Luo, C.; Li, Y. Acta Pharm. Sin. 2010, 45, 1017 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-YXXB201008017.htm
Fu, X.-Z.; Ou, Y.; Pei, J.-Y.; Liu, Y.; Li, J.; Zhou, W.; Lan, Y.-Y.; Wang, A.-M.; Wang, Y.-L. Eur. J. Med. Chem. 2012, 49, 211. doi: 10.1016/j.ejmech.2012.01.013
Magee, W. C.; Evans, D. H. Antiviral Res. 2012, 96, 169. doi: 10.1016/j.antiviral.2012.08.010
(a) De Clercq, E. Trends Pharmacol. Sci. 2002, 23, 456.
(b) Andrei, G.; Topalis, D.; De Schutter, T.; Snoeck, R. Antiviral Res. 2015, 114, 21.
Cundy, K. C.; Bidgood, A. M.; Lynch, G.; Shaw, J. P.; Griffin, L.; Lee, W. A. Drug. Metab. Dispos. 1996, 24, 745.
(a) Beadle, J. R.; Hartline, C.; Aldern, K. A.; Rodriguez, N.; Harden, E.; Kern, E. R.; Hostetler, K. Y. Antimicrob. Agents Chemother. 2002, 46, 2381.
(b) Hostetler, K. Y. Viruses 2010, 2, 2213.
Marty, F. M.; Winston, D. J.; Rowley, S. D.; Vance, E.; Papanicolaou, G. A.; Mullane, K. M.; Brundage, T. M.; Robertson, A. T.; Godkin, S.; Mommeja-Marin, H. N. Engl. J. Med. 2013, 369, 1227. doi: 10.1056/NEJMoa1303688
Marty, F. M.; Winston, D. J.; Chemaly, R. F.; Boeckh, M. J.; Mullane, K. M.; Shore, T. B.; Papanicolaou, G. A.; Morrison, M. E.; Brundage, T. M.; Mommeja-Marin, H. Biol. Blood Marrow. Transplant. 2016, 22, S23. http://xueshu.baidu.com/usercenter/paper/show?paperid=426e332490b2d62194312dad04a47e02&site=xueshu_se
(a) Florescu, D.; Grimley, M.; Papanicolaou, G.; Prasad, V.; Vainorius, E.; Chittick, G.; Brundage, T.; Nichols, G. Am. J. Transplant. 2018, 18, S360.
(b) Chittick, G.; Morrison, M.; Brundage, T.; Nichols, W. G. Antivir. Res. 2017, 143, 269.
Dunning, J.; Kennedy, S. B.; Antierens, A.; Whitehead, J.; Ciglenecki, I.; Carson, G.; Kanapathipillai, R.; Castle, L.; Howell-Jones, R.; Pardinaz-Solis, R. PLoS One 2016, 11, e0162199. doi: 10.1371/journal.pone.0162199
Lampertico, P. Gut 2014, 63, 869. doi: 10.1136/gutjnl-2013-305859
Mak, L.-Y.; Seto, W.-K.; Lai, C.-L.; Yuen, M.-F. Expert Opin. Drug Metab. Toxicol. 2018, 14, 101. doi: 10.1080/17425255.2018.1417983
Ahn, S. H.; Kim, W.; Jung, Y. K.; Yang, J. M.; Jang, J. Y.; Kweon, Y. O.; Cho, Y. K.; Kim, Y. J.; Hong, G. Y.; Kim, D. J. J. Hepatol. 2017, 66, S88.
Lai, C.-L.; Ahn, S. H.; Lee, K. S.; Um, S. H.; Cho, M.; Yoon, S. K.; Lee, J.-W.; Park, N. H.; Kweon, Y.-O.; Sohn, J. H. Gut 2014, 63, 996. doi: 10.1136/gutjnl-2013-305138
Yuen, M.-F.; Ahn, S. H.; Lee, K. S.; Um, S. H.; Cho, M.; Yoon, S. K.; Lee, J.-W.; Park, N. H.; Kweon, Y. O.; Sohn, J. H. Hepatology 2013, 58, 693A.
Yuen, M.-F.; Ahn, S. H.; Lee, K. S.; Um, S. H.; Cho, M.; Yoon, S. K.; Lee, J.-W.; Park, N. H.; Kweon, Y.-O.; Sohn, J. H. J. Hepatol. 2015, 62, 526. doi: 10.1016/j.jhep.2014.10.026
Baszczynski, O. Janeba, Z. Med. Res. Rev. 2013, 33, 1304. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM23893552
Luo, M.; Groaz, E.; Andrei, G.; Snoeck, R.; Kalkeri, R.; Ptak, R. G.; Hartman, T.; Buckheit, R. W., Jr.; Schols, D.; De Jonghe, S. J. Med. Chem. 2017, 60, 6220. doi: 10.1021/acs.jmedchem.7b00416
Holy, A.; Gunter, J.; Dvorakova, H.; Masojidkova, M.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E. J. Med. Chem. 1999, 42, 2064. doi: 10.1021/jm9811256
Jansa, P.; Baszczyňski, O.; Dračínský, M.; Votruba, I.; Zídek, Z.; Bahador, G.; Stepan, G.; Cihlar, T.; Mackman, R.; Holý, A; Jansa, Z. Eur. J. Med. Chem. 2011, 46, 3748. doi: 10.1016/j.ejmech.2011.05.040

图 1 第一代ANPs代表药物及其已上市前药
Figure 1 The first generation ANPs drugs and their marketed prodrugs

图 4 ADV肝靶向磷酸酯前药和氨基酸前药
Figure 4 Phospholipid liver targeting prodrug and amino acid ester prodrug of TFV

图 6 引入非甾体抗炎药和L-氨基酸的阿德福韦前药
Figure 6 Nonsteroidal anti-inflammatory agents and amino acids conjugates of TFV

图 8 鸟嘌呤核苷膦酸酯型前药及其转化
Figure 8 Guanine nucleoside phosphonate prodrugs and the conversion of 23 to 25

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:






