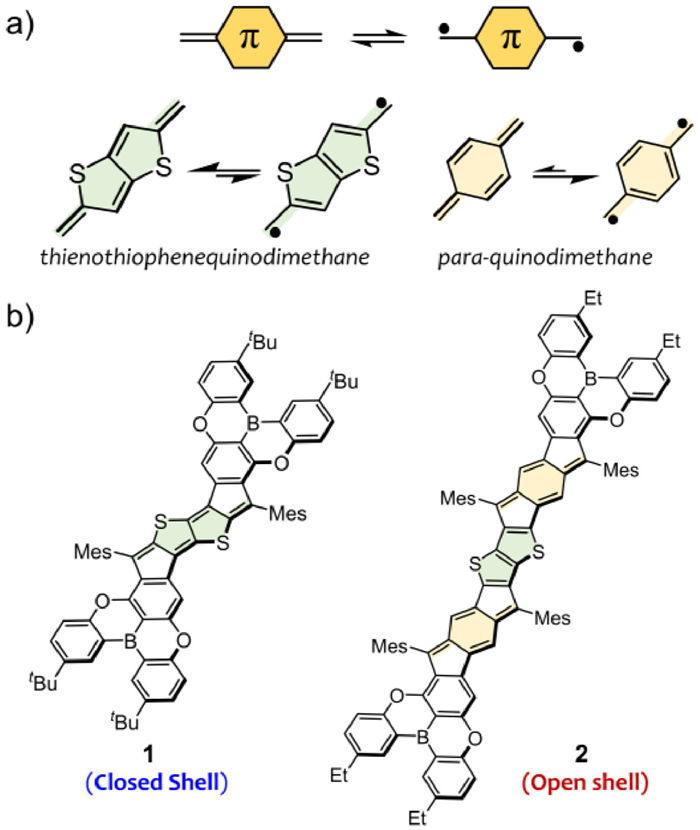
Figure 1.
(a) Two representative quinoidal π-subunits and their resonance structures. (b) Chemical structures of thienothiophene-embedded ladder-type molecules (1 and 2). Mes = mesityl.
Thienothiophene-centered ladder-type π-systems that feature distinct quinoidal π-extension
Jiaxiang Guo , Zeyi Li , Tianyu Zhang , Xinyu Tian , Yue Wang , Chuandong Dou

Quinoidal π-conjugated structures are important fundamental subunits for organic π-systems. They may possess a resonance mixed state of closed-shell and open-shell forms, induce narrow energy gap and even diradical characteristic, and thereby produce some intriguing optical, electronic and magnetic properties of polycyclic hydrocarbons (PHs) [1-11]. Quinodimethanes (QDMs), one kind of the simplest pro-aromatic diradicals, have been intensively used as quinoidal subunits to construct diradical and polyradical PHs (Fig. 1a). Till now, a variety of QDM-containing PHs with one-, two- and three-dimensional topologies and planar and nonplanar conformations have been developed, and they exhibited great potential for applications in organic electronics and spintronics [12-20]. Quinoidal thienoacenes (QTAs) have been employed as another kind of π-subunits to construct the diindenothienoacene derivatives for understanding (anti)aromaticity and developing optical materials [21-25]. Nevertheless, the comparative study on QDMs and QTAs is still scarce, and moreover, their distinct effects on electronic structures and physical properties of π-systems remain unclear. Thus, the in-depth study of these quinoidal π-subunits is of importance for (anti)aromatic chemistry and π-electron materials.
Incorporation of heteroatoms (e.g., boron, nitrogen and sulfur) into PHs is a powerful approach to modulate open-shell characteristics [26-33]. We have recently made efforts at the development of boron-containing organic diradicaloids. Based on our proposed design strategy, namely the borylation of antiaromatic PHs, we developed B/O-type and B/N-type organic diradicaloids, disclosed the heteroatom-doping effects on the electronic structures and physical properties of PHs, and achieved high photothermal conversion capability [34-38]. These open-shell conjugated organoboranes thus provide a potential scaffold to design thienoacene-embedded π-systems for gaining insight into the electronic effects of QDMs and QTAs on PHs.
Herein, we designed and synthesized two thienothiophene-centered ladder-type polycyclic molecules 1 and 2, which feature one quinoidal thienothiophene (QTT) moiety and two para-quinodimethane (p-QDM) subunits, respectively (Fig. 1b). Such distinct quinoidal π-extension modes lead to their different electronic structures and properties. Notably, while 1 is a fully closed-shell molecule, 2 possesses an open-shell structure along with partial contribution of tetraradical state. Moreover, although 2 has a larger π-conjugated skeleton and open-shell electronic state, it exhibits enlarged bandgap and blue-shifted absorption. Herein, we disclose their synthesis, (anti)aromatic structures and electrochemical and photophysical properties, together with in-depth investigation of the dication of 1.
The target compounds 1 and 2 were synthesized in two different synthetic routes (Scheme 1). The Suzuki‒Miyaura cross-coupling reaction between 3 and 3,6-dibromothieno[3,2-b]thiophene-2,5-dicarbaldehyde afforded the dialdehyde 5. The nucleophilic addition reaction on 5 with mesityl magnesium bromide and then intramolecular Friedel-Crafts alkylation reaction on the diol intermediate with BF3·Et2O produced the tetrahydro-precursor 6. The final oxidative dehydrogenation reaction was performed on 6 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to obtain the target molecule 1. In the synthesis of 2, the Suzuki‒Miyaura cross-coupling and then Stille coupling produced the precursor 7 that contains four mesitylketone groups [33]. The subsequent reduction of 7 with LiAlH4, Friedel‒Crafts cyclization on the tetraol species and final oxidative dehydrogenation reaction on 8 afforded 2. 1 and 2 were purified using silica gel column chromatography, and their formations were determined using 1H NMR spectroscopy and high-resolution mass spectrometry (HRMS) (Fig. S1 in Supporting information). In their 1H NMR spectra, the CDCl3 solution of 1 exhibits the full aryl proton signals at room temperature, whereas the aryl proton signals for 2 are not fully observed (Figs. S14 and S18 in Supporting information). This preliminary result indicates that 1 and 2 are closed-shell and open-shell molecules (vide infra), respectively.

We first performed cyclic voltammetry measurement on 1 to study its redox activity. The measurement was conducted on its CH2Cl2 solution using n-Bu4NPF6 (0.1 mol/L) as supporting electrolyte (Fig. 2a). It displays two reversible oxidation processes with half-wave potentials (Eox) at +0.29/+0.57 V (versus Fc/Fc+), as well as two irreversible reduction processes with potentials (Ered) at −1.34/−1.60 V determined by the peaks in differential pulse voltammogram (DPV). Its reversible oxidation activity suggests that 1 can be facilely oxidized into stable radical cation and dication.
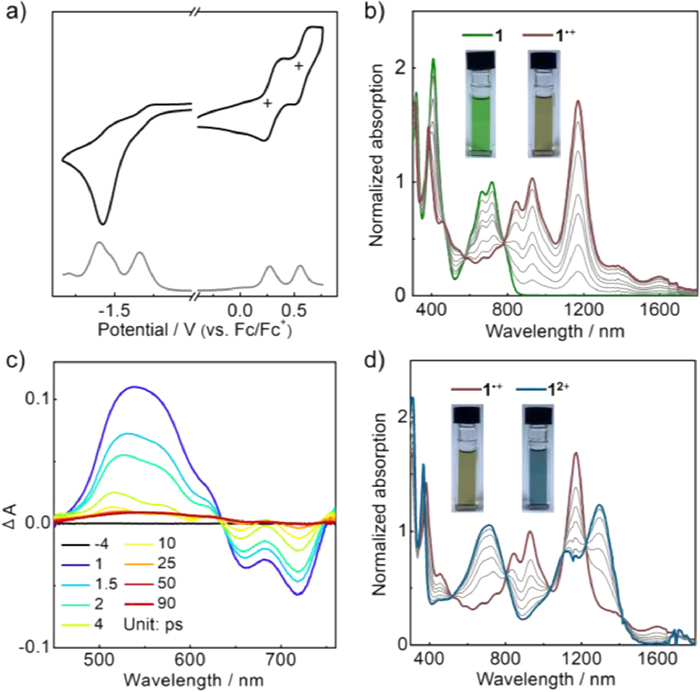
In the UV–vis-NIR absorption spectrum, the CH2Cl2 solution of 1 exhibits two main absorption bands around 710 and 410 nm (Fig. 2b). The absorption peaks (λabs) in the low-energy region are observed at 716/665 nm, along with a weak shoulder around 790 nm (Fig. S3 in Supporting information). Based on the onset absorption, the optical bandgap (Egopt) is estimated to be 1.47 eV. The absorption spectrum is well reproduced by the time-dependent DFT (TD-DFT) calculation on 1′ at the B3LYP/6–311G(d) level (Figs. S7 and S9 in Supporting information). The low-energy absorption band at 710 nm is assigned to the transition of S0 → S3, and its shoulder peak is attributable to the forbidden S0 → S2 transition. Both of these two transitions involve the LUMO of 1′, which is well delocalized over its quinoidal π-skeleton.
The excited-state dynamics of 1 were investigated by femtosecond transient absorption (fs-TA) spectroscopy. The fs-TA spectrum of 1 in toluene displays intense excited-state absorption (ESA) bands in a range of 450−635 nm and two ground-state bleach (GSB) peaks at 718/662 nm (Fig. 2c), which match well with their steady-state absorption spectra. The decay profile probed at 660 nm is fitted following double exponential function, determining the singlet excited-state lifetimes (τ) of 12.2 and 1.5 ps (Fig. S4 in Supporting information). Such short singlet excited-state lifetimes are indicative of the ultrafast relaxation process and non-fluorescence nature, which is usually observed in most antiaromatic π-systems [23,39,40].
Stepwise oxidation titration of 1 using NO·SbF6 afforded its radical cation 1•+ and dication 12+, and this progress was monitored by UV–vis-NIR absorption spectroscopy. Upon addition of the first equivalent NO·SbF6 to a CH2Cl2 solution of 1, the absorption peaks of 1 at 716/665 nm decrease, and the new absorptions of 1•+appear at 1172/928/842 nm with a long-wavelength tail at 1598/1378 nm (Fig. 2b). Upon addition of the second equivalent NO·SbF6, the absorption of 1•+ at 1172 nm gradually decays, and a near-infrared absorption band of 12+emerges at 1294/1121 nm (Fig. 2d). According to the TD-DFT calculation at the B3LYP/6–311+G(d) level, this absorption band is attributed to the transition of S0 → S1 of 1′2+ (Fig. S10 in Supporting information). This transition involves the HOMO and HOMO-7 that are delocalized over the whole molecules, thus revealing the remarkable contribution of the oxygen atoms of 12+ (vide infra).
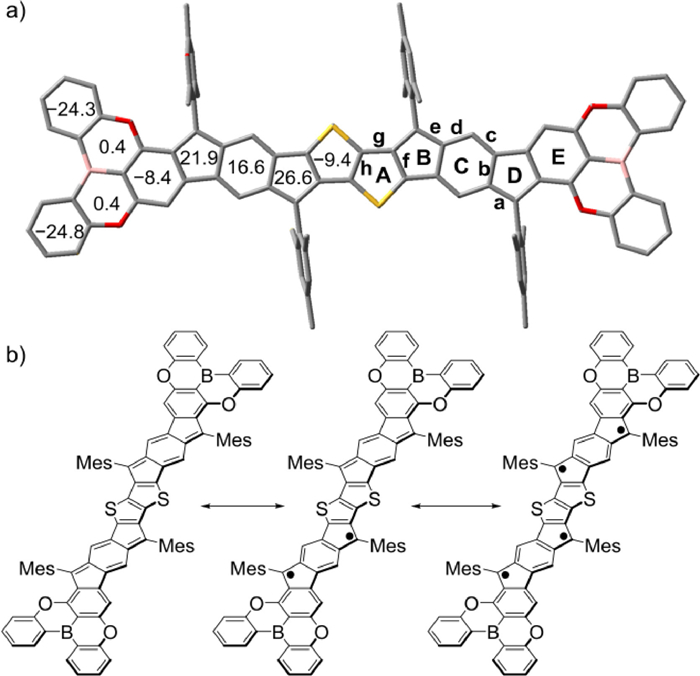
Single crystals suitable for X-ray crystallographic analysis were grown by diffusion of methanol into the CH2Cl2 solution of 1. As shown in Fig. 3a, 1 has an almost planar centrosymmetric framework along with two helicene-like terminal oxygen-bridged triphenylborane groups. The bond-length analysis shows that the C–C bond lengths in the central thiophenothiophene core is 1.368 Å for bond a, 1.442 Å for bond b, 1.359 Å for bond c and 1.444 Å for bond d (Fig. S5 in Supporting information). This large bond-length alternation is in consistent with the single-double bond alternation pattern in resonance form of 1 (Fig. 3c), indicating its characteristic quinoidal conjugated structure. To investigate the (anti)aromaticity of the ladder π-system, nucleus-independent chemical shift (NICS) calculation was performed on 1′ (Fig. 3d) at the GIAO-UB3LYP/6–311+G(d) level of theory. The large positive NICS(1)ZZ value of ring B confirms the antiaromaticity of thiophenothiophene core with quinoidal structure, and the negative NICS(1)ZZ value of ring C indicates that it is an aromatic ring. These results are in accordance with the bond-length analysis of single-crystal structure. Thus, the closed-shell quinoidal resonance form mainly contributes to the ground state of 1 rather than the open-shell diradical one.
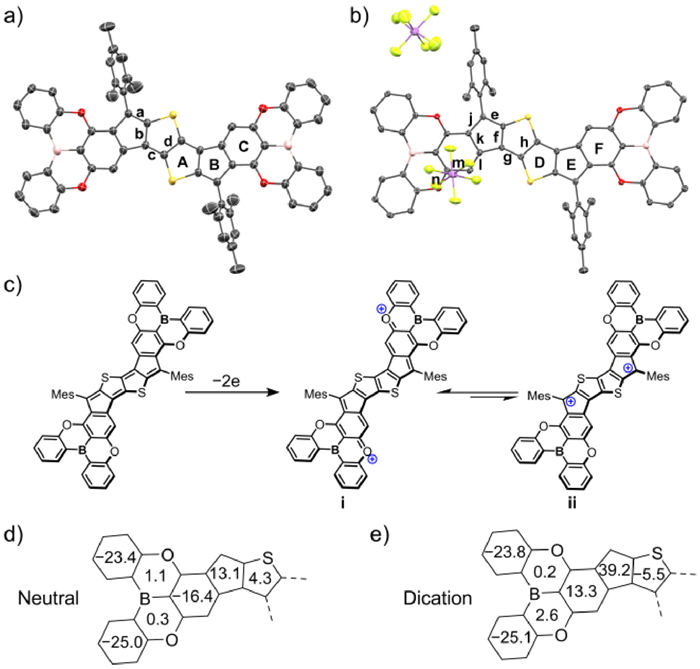
Single crystals of 12+ were obtained via the diffusion of n-hexane into its CH2Cl2 solution under argon. 12+ owns the similar conformation to 1 (Fig. 3b). Notably, in comparison to 1,12+ shows the shorter bond lengths (1.389/1.418 Å) for bonds f/h and the longer bond length (1.397 Å) for bond g, implying the aromatic pattern of the thiophenothiophene core (Fig. S6 in Supporting information). In addition, 12+ exhibits the shortened bond length of 1.407 Å for bond j and 1.336 Å for bond n, indicative of the quinoidal pattern of ring F. The NICS calculation (Fig. 3e) shows the negative NICS(1)ZZ value for ring D and positive NICS(1)ZZ values of ring E and F, thus demonstrating the aromaticity of the thiophenothiophene core and antiaromaticity of ring E and F in 12+ [41,42]. Accordingly, the resonance form i in Fig. 3c mainly contributes to the ground state of 12+, whose positive charges are delocalized onto the oxygen atoms. This analysis is in accordance with the DFT calculation results of 1′2+.
To reveal the open-shell nature of 2, we performed variable-temperature (VT) 1H NMR measurements on its CDCl3 solution. At 293 K, the solution exhibits partially broadened signals, which gradually sharpen and increase in intensity when decreasing the temperature (Fig. 4a). When the temperature is decreased to 223 K, a new peak at 7.69 ppm emerges and well-resolved signals of all protons in the aromatic region are observed. These spectral changes are indicative of the reduced thermally excited triplet species of 2 at lower temperature, which is typical for PHs with open-shell singlet ground state. Thus, 2 is an open-shell molecule with singlet ground state.
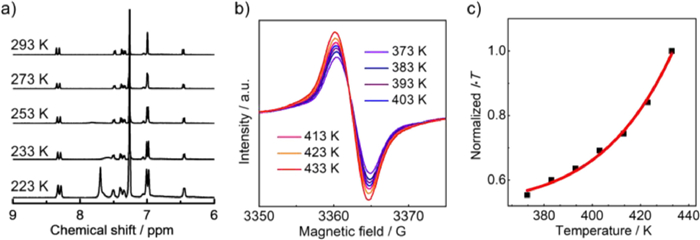
The VT electron paramagnetic resonance (VT-EPR) measurements were performed on 2 to gain insight into the open-shell character and magnetic properties. The solid powder of 2 exhibits the unresolved EPR signals at the temperature range of 373‒433 K with g-factor of 2.0036 (Fig. 4b). The signal intensity gradually increases with enhancing the temperature, which is attributed to the increase of thermally accessible triplet species at higher temperature. This behavior reveals the singlet open-shell ground state of 2. By fitting I·T versus T through the Bleaney–Bowers equation, the singlet-triplet energy gap (ΔES–T) is calculated to be −11.34 kcal/mol (Fig. 4c). The relatively large ΔES–T suggests the moderate open-shell extent of 2.
To explore the electronic structures in terms of open-shell nature and (anti)aromaticity, theoretical studies were carried out on the model molecule 2′. Density functional theory (DFT) calculations were performed at the UB3LYP/6–311G(d) level of theory. The optimized geometry of 2′ possesses a centrosymmetric structure (Fig. 5). The bond lengths of bond a/e are 1.391/1.392 Å, which are longer than typical double bond length (~1.33 Å), implying these two p-QDMs exist as an open-shell state to some extents. Thus, 2 has an open-shell electronic state along with partial tetraradical contribution, which is further proved by the spin density distribution of 2′ (Fig. S8 in Supporting information). Based on the occupation numbers of the spin-unrestricted Hartree-Fock natural orbitals (UHF-UB3LYP/6–311G(d)), the diradical and tetraradical index (y0 and y1) are calculated to be 0.33 and 0.31, confirming the singlet open-shell nature of 2 [43]. In addition, the theoretical ΔES−T value of 2′ is −13.96 kcal/mol, which is similar to the experimental value of 2. On the other hand, the negative NICS(1)ZZ value of ring A reveals the aromaticity of the thiophenothiophene core, whereas the positive NICS(1)ZZ values of ring B, C and D indicate their antiaromaticity that is ascribed to the p-QDM subunit in 2. It means that the aromatic thiophenothiophene core separates these two p-QDM subunits into independent quinoidal substructures. As a result, they may contribute to the open-shell resonance forms individually, and thus 2 has the nearly identical diradical and tetraradical indexes. It can be concluded that the π-extension of and embedded p-QDM subunits in 2 induce its open-shell ground state.
We then performed the UV–vis-NIR absorption measurement on 2 to study its photophysical properties. As shown in Fig. 6a, two main absorption bands are located at 664 and 464 nm, along with an absorption tail that extends over 850 nm. According to the TD-DFT calculation of 2′, the absorption band around 664 nm is assigned to the transitions of S0 → S7 and S0 → S11 (Fig. S11 in Supporting information). Moreover, the optical bandgap of 2 is estimated to be 1.79 eV based on the onset absorption. Generally, larger π-conjugated skeleton and open-shell electronic state can lead to more red-shifted absorption spectra and smaller energy gap. From 1 to 2, the main absorption peak is blue-shifted by 52 nm and the optical bandgap is enlarged by 0.32 eV. These unexpected changes can be attributed to the broken quinoidal conjugation by the aromatic thiophenothiophene in 2. Namely, 2 has two separated p-QDM substructures that involve three rings, respectively, whereas 1 has a larger quinoidal structure involving four rings and thus it has the red-shifted absorption and smaller bandgap. This finding reveals that rational fusion of p-QDM can produce open-shell electronic structure but with large energy gap of PHs.
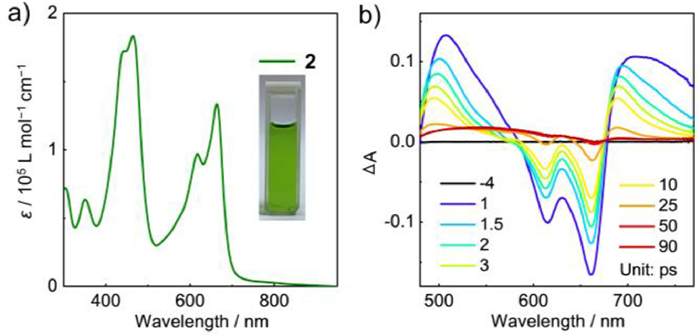
The femtosecond transient absorption spectra of 2 in toluene show the broad excited-state absorption bands at 480–580 nm and 680–770 nm (Fig. 6b). The ground-state bleach band appear at 580–680 nm, along with the peaks at 660/615 nm, which match well with their low-energy absorption signals of the steady-state absorption spectra. The singlet excited-state lifetimes of 2 are determined to be 14.2 and 0.9 ps. These short excited-state lifetimes indicate the fast relaxation for the singlet excited states that are usually reported for antiaromatic π-systems and radical PHs [23,39,40].
In conclusion, two thienothiophene-centered ladder-type polycyclic molecules (1 and 2) were synthesized, which possess one quinoidal thienothiophene moiety and two p-QDM subunits, respectively. As theoretically and experimentally studied, while 1 is a fully closed-shell molecule, 2 owns an open-shell structure along with partial contribution of tetraradical state. Moreover, although 2 has a larger π-conjugated skeleton and open-shell electronic state, it exhibits larger bandgap and blue-shifted absorption. On the other hand, the reversible oxidation activity of 1 enables the preparation of its dication 12+, and the studies on its single-crystal and aromatic structures demonstrate that its two positive charges are delocalized onto the oxygen atoms, thus achieving fully π-extended structure and near-infrared absorption. This study not only gains insight into quinoidal π-subunits, but also provides an important basis for the development of antiaromatic and open-shell functional π-electron materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by National Natural Science Foundation of China (Nos. 22175074 and 52373182) and Jilin Scientific and Technological Development Program (No. 20220101054JC).
Supplementary material associated with this article can be found, in the online version, at doi:
Z.X. Chen, Y. Li, F. Huang, Chem 7 (2021) 288–332. doi: 10.1016/j.chempr.2020.09.024
W. Zeng, J. Wu, Chem 7 (2021) 358–386. doi: 10.1016/j.chempr.2020.10.009
X. Hu, W. Wang, D. Wang, et al., J. Mater. Chem. C 6 (2018) 11232–11242. doi: 10.1039/C8TC04484H
J.X. Dong, H.L. Zhang, Chin. Chem. Lett. 27 (2016) 1097–1104. doi: 10.1016/j.cclet.2016.05.005
T. Kubo, Chem. Rec. 15 (2015) 218–232. doi: 10.1002/tcr.201402065
L. Wang, X. Shi, S. Feng, et al., CCS Chem. 4 (2022) 2748–2756. doi: 10.31635/ccschem.021.202101199
T. Du, R. Gao, Y. Deng, et al., Angew. Chem. Int. Ed. 59 (2020) 221–225. doi: 10.1002/anie.201911530
C. Wang, Y. Yang, L. Lin, et al., Angew. Chem. Int. Ed. 62 (2023) e202307856. doi: 10.1002/anie.202307856
S. Qiu, Y. Zhao, L. Zhang, et al., CCS Chem. 5 (2023) 1763–1772. doi: 10.31635/ccschem.023.202302830
Y. Zeng, R. Duan, Y. Guo, et al., Chin. Chem. Lett. 30 (2019) 211–216. doi: 10.1016/j.cclet.2018.05.029
A. Obolda, M. Zhang, F. Li, Chin. Chem. Lett. 27 (2016) 1345–1349. doi: 10.1016/j.cclet.2016.06.030
Y. Tobe, Top Curr. Chem. 376 (2018) 12. doi: 10.1007/s41061-018-0189-0
J. Ma, J. Liu, M. Baumgarten, et al., Angew. Chem. Int. Ed. 56 (2017) 3280–3284. doi: 10.1002/anie.201611689
G.E. Rudebusch, J.L. Zafra, K. Jorner, et al., Nat. Chem. 8 (2016) 753–759. doi: 10.1038/nchem.2518
W. Zeng, Z. Sun, T.S. Herng, et al., Angew. Chem. Int. Ed. 55 (2016) 8615–8619. doi: 10.1002/anie.201602997
J. Guo, Z. Li, J. Zhang, et al., CCS Chem. 4 (2022) 95–103. doi: 10.31635/ccschem.021.202000658
R.Q. Lu, S. Wu, L.L. Yang, et al., Angew. Chem. Int. Ed. 58 (2019) 7600–7605. doi: 10.1002/anie.201902028
H. Han, D. Zhang, Z. Zhu, et al., J. Am. Chem. Soc. 143 (2021) 17690–17700. doi: 10.1021/jacs.1c08262
L. Ma, S. Wang, Y. Li, et al., CCS Chem. 4 (2022) 3669–3676. doi: 10.31635/ccschem.022.202201954
F. Lombardi, A. Lodi, J. Ma, et al., Science 366 (2019) 1107–1110. doi: 10.1126/science.aay7203
C. Zhang, X. Zhu, Acc. Chem. Res. 50 (2017) 1342–1350. doi: 10.1021/acs.accounts.7b00050
W. Liu, W. Li, J. Yao, et al., Chin. Chem. Lett. 29 (2018) 381–384. doi: 10.1016/j.cclet.2017.11.018
X. Shi, P.M. Burrezo, S. Lee, et al., Chem. Sci. 5 (2014) 4490–4503. doi: 10.1039/C4SC01769B
G.E. Rudebusch, A.G. Fix, H.A. Henthorn, et al., Chem. Sci. 5 (2014) 3627–3633. doi: 10.1039/C4SC01432D
Q. Wu, R. Li, W. Hong, et al., Chem. Mater. 23 (2011) 3138–3140. doi: 10.1021/cm201326c
Z. Feng, S. Tang, Y. Su, et al., Chem. Soc. Rev. 51 (2022) 5930–5973. doi: 10.1039/D2CS00288D
L. Mao, M. Zhou, X. Shi, et al., Chin. Chem. Lett. 32 (2021) 3331–3341. doi: 10.1016/j.cclet.2021.05.004
T. Kushida, S. Shirai, N. Ando, et al., J. Am. Chem. Soc. 139 (2017) 14336–14339. doi: 10.1021/jacs.7b05471
L. Yuan, J. Guo, Y. Yang, et al., CCS Chem. 5 (2023) 876–884. doi: 10.31635/ccschem.022.202101738
Z.Y. Wang, Y.Z. Dai, L. Ding, et al., Angew. Chem. Int. Ed. 60 (2021) 4594–4598. doi: 10.1002/anie.202012989
J.E. Barker, J.J. Dressler, A.C. Valdivia, et al., J. Am. Chem. Soc. 142 (2020) 1548–1555. doi: 10.1021/jacs.9b11898
J.J. Dressler, M. Teraoka, G.L. Espejo, et al., Nat. Chem. 10 (2018) 1134–1140. doi: 10.1038/s41557-018-0133-5
W. Wang, F. Hanindita, R.D. Webster, et al., CCS Chem. 5 (2023) 1108–1117. doi: 10.31635/ccschem.022.202202165
J. Guo, X. Tian, Y. Wang, et al., Chem. Res. Chin. Univ. 39 (2023) 161–169. doi: 10.1007/s40242-023-2363-3
J. Guo, Y. Yang, C. Dou, et al., J. Am. Chem. Soc. 143 (2021) 18272–18279. doi: 10.1021/jacs.1c08486
T. Sun, J. Guo, H. Wen, et al., Aggregate 4 (2023) e362. doi: 10.1002/agt2.362
J. Guo, Z. Li, X. Tian, et al., Angew. Chem. Int. Ed. 62 (2023) e202217470. doi: 10.1002/anie.202217470
Z. Li, T. Sun, J. Guo, et al., Org. Chem. Front. 10 (2023) 4289–4297. doi: 10.1039/D3QO00900A
Z. Zeng, S. Lee, M. Son, et al., J. Am. Chem. Soc. 137 (2015) 8572–8583. doi: 10.1021/jacs.5b04156
P. Hu, S. Lee, T.S. Herng, et al., J. Am. Chem. Soc. 138 (2016) 1065–1077. doi: 10.1021/jacs.5b12532
K.B. Wiberg, Chem. Rev. 101 (2001) 1317–1331. doi: 10.1021/cr990367q
Y. Liu, L. Yuan, J. Guo, et al., Angew. Chem. Int. Ed. 62 (2023) e202306911. doi: 10.1002/anie.202306911
A. Rana, Y. Hong, T.Y. Gopalakrishna, et al., Angew. Chem. Int. Ed. 130 (2018) 12714–12717. doi: 10.1002/ange.201807411

Figure 1 (a) Two representative quinoidal π-subunits and their resonance structures. (b) Chemical structures of thienothiophene-embedded ladder-type molecules (1 and 2). Mes = mesityl.

Scheme 1 Synthesis of 1 and 2. Reagents and conditions: (a) Pd(PPh3)4, Na2CO3, toluene: ethanol: H2O (5:2:1), 60 ℃. (b) Pd(PPh3)4, K2CO3, toluene: ethanol: H2O (5:1:1), 70 ℃. (c) (i) Mesityl magnesium bromide, THF, 25 ℃; (ii) BF3·Et2O, CH2Cl2, 25 ℃. (d) DDQ, toluene, 80 ℃. (e) Pd(PPh3)4, toluene, 110 ℃. (f) (i) LiAlH4, THF, 50 ℃; (ii) BF3·Et2O, CH2Cl2, 25 ℃.

Figure 2 (a) Cyclic voltammograms of 1 in CH2Cl2 at a scan rate of 100 mV/s. (b) UV–vis-NIR absorption spectra of 1 and 1•+ in CH2Cl2, along with the oxidation process using NOSbF6. Inset are the photographs. (c) Femtosecond transient absorption (fs-TA) spectra of 1 in toluene with photoexcitation at 400 nm. (d) UV–vis-NIR absorption spectra of 1•+ and 12+ in CH2Cl2, along with the oxidation process using NOSbF6. Inset are the photographs.

Figure 3 X-ray single-crystal structures of (a) 1 and (b) 12+ (50% probability for thermal ellipsoids). The tBu groups are omitted for clarity. (c) The oxidation process from 1′ to 1′2+, and the resonance structures of 1′2+. Calculated NICS(1)ZZ values of (d) 1′ and (e) 1′2+.

Figure 4 (a) Variable-temperature 1H NMR spectra of 2 in CDCl3. (b) Variable-temperature EPR spectra of 2 in the solid state. (c) Experimental (black dots) and Bleaney-Bowers fit (red line) of I·T − T plots.

Figure 5 (a) Optimized structure and calculated NICS(1)ZZ values of 2′. (b) The closed-shell and open-shell resonance structures of 2′.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
