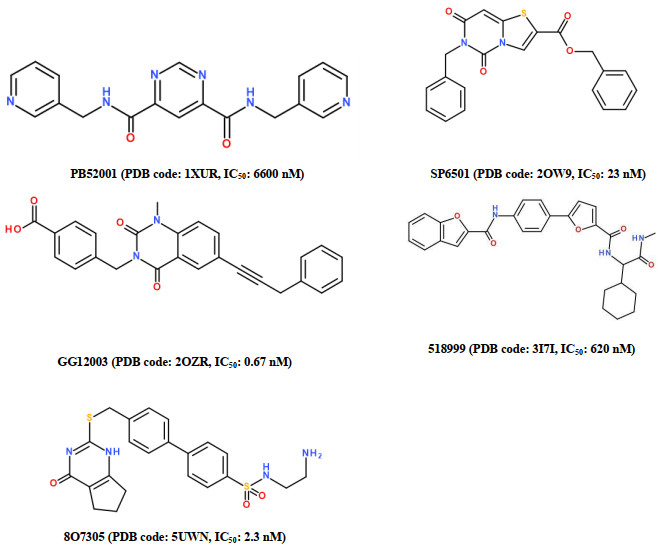
Figure 1.
Molecular structures of five ligands involved in the five experimental MMP-13-ligand complexes and the IC50 values
Structure-based Screening for the Non-zinc-chelating Selective MMP-13 Inhibitors of Natural Products
Qing ZOU , Qiu-Shuang GAO , Peng YAO , Qi-Zheng YAO , Ji ZHANG

Matrix metalloproteinases (MMPs) are a significant family of zinc-dependent endopeptidases. MMPs have more than 20 members that are involved in the degradation of extracellular matrix (ECM) during cellular migration and tissue remodeling[1]. Over-expression of MMPs can break the balance and cause the excessive degradation of ECM, resul- ting in cancer, arthritis, atherosclerosis, and periodontal disease[2, 3]. Among MMPs, MMP-13 has been considered as a promising therapeutic target for osteoarthritis (OA). OA is a degenerative joint disease characterized by growing loss of articular cartilage, which results in chronic pain and reduced mobility[4, 5]. In the experimental OA models, the expression pattern of MMP-13 correlates with the presence of patholo- gical chondrocytes that undergo hypertrophic differentiation in the early stage of OA development[6]. However, the clinical utilities of broad-spectrum MMP inhibitors, mostly hydro- xamic acids, have been restricted by the painful and joint-stiffening side effects described as musculoskeletal syndrome (MSS). It is believed that MSS is generated by the nonselective inhibition of multiple and critical MMPs or the powerful chelating ability between hydroxamic acids and the catalytic zinc ions of various MMPs[7, 8]. On the other hand, the differences of S1′ pockets between MMP family members provide an opportunity to achieve the selective binding of inhibitors with MMPs[9]. In particular, MMP-13 possesses the largest S1′ pocket among MMPs, and such a S1′ pocket is found to be flexible and not observed in the other MMPs. Besides, The S1' pockets of various MMPs do not share the
similarity of amino acid sequence. Therefore, the S1′ pocket of MMP-13 has been considered to be a specific target for designing the selective inhibitors that do not chelate the catalytic zinc ion of MMP-13, i.e., designing the selective non-zinc-chelating MMP-13 inhibitors[10-19].
In drug studies, natural products are usually used as lead compounds because of their physicochemical diversity, lower cytotoxicity, and greater therapeutic efficiency, compared with synthetic drug-like compounds[20, 21]. Besides, pharmacophore modeling and molecular docking are efficient computational approaches in drug discovery because they are cost-effective and time saving[22, 23]. In this study, the pharmacophore model of MMP-13 inhibitors was first built based on the five experimental crystal structures of receptor-ligand complexes, which involves the interactions of inhibitors with the S1' pocket of MMP-13, but the inhibitors do not chelate the catalytic zinc ion of MMP-13. Then, a decoy set was employed to validate the reliability of pharmacophore modeling. Next, the screening was performed based on both pharmacophore modeling and molecular docking. Finally, the four selective non-zinc-chelating MMP-13 inhibitors of natural products were obtained, and their binding modes with MMP-13 receptor were further discussed.
In RCSB Protein Data bank (PDB), we have found the 37 experimental crystal structures of MMP-13-ligand complexes[9]. To build the reliable pharmacophore modeling, we selected five of these 37 experimental crystal structures, including PDB codes: 1XUR, 2OW9, 2OZR, 3I7I, and 5UWN, where each ligand can well interact with the S1' pocket of MMP-13 and not chelate the catalytic zinc ion of MMP-13[10-14], indicating that these ligands are the selective non-zinc-chelating MMP-13 inhibitors. These five crystal complexes were prepared using the Prepare Protein protocol in Discovery Studio 3.0 (DS 3.0) program[24], to correct the structure problems due to nonstandard naming, structural disorder, protein residue, and missing side-chain or backbone atoms. Fig. 1 shows the molecular structures of five ligands that were extracted from these five MMP-13-ligand complexes, with their corresponding IC50 values.
Using the above five MMP-13-ligand complexes, we built one multiple structure-based pharmacophore model by employing the Receptor-Ligand Pharmacophore Generation (RLPG) protocol in DS 3.0. The RLPG protocol has the following notable features: (i) it uses adjustable shape constraints to determine the interactions between the binding ligand and protein, (ii) it creates all possible pharmacophore combinations and returns the best one according to ranking scores and best fit values. In our calculations, a set of standard pharmacophore features were considered, including hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), positive ionizable (PI), negative ionizable (NI), hydrophobic (HY), and ring aromatic (RA). Excluded volumes were added at the conserved zinc catalytic site of MMP-13 for avoiding those ligands to chelate the catalytic zinc ion in the MMP-13 receptor. According to the structures of imported receptors and ligands, the several good initial queries generated. These queries were then aligned and clustered by the Cluster protocol to define the center features. Finally, one pharmaco- phore model was built based on these five crystal structures of MMP-13-ligand complexes.
The purpose of validating a pharmacophore model is to verify whether it is able to identify active molecules from many molecules. Here, we built a decoy set to test the performance of pharmacophore model. This decoy set contains 20 known active molecules and 1200 known inactive molecules, and they were collected from Binding Database (www.bindingsb.org) where all the molecules were obtained from experimental results. The Ligand Pharmacophore Mapping protocol was employed to map the decoy molecules to the pharmacophore model. According to the screening results, the enrichment factor (EF)[25], Güner-Henry (GH) scores, and receiver operating characteristic (ROC) curve were used to assess the quality of built pharmacophore model[26]. The relevant equations are shown below:
|
|
|
|
Where D and A are the total number of molecules and the number of active molecules in a decoy set, respectively. Ht is the number of compounds that pharmacophore hits, and Ha is the number of actives in the hit list named true positives.
AnalytiCon Discovery and InterBioScreen are the two world-wide leading providers for the pure natural products, and now there have been 5154 and 56799 natural products in their databases, respectively. In our screening, all the compounds in these two databases were first filtered through Lipinski's rule of five to ensure the drug-like properties. Then, the validated pharmacophore model was used to screen the obtained drug-like compounds, based on the fast flexible searching algorithm, as implemented in Ligand Pharmaco- phore Mapping protocol in DS 3.0. The compounds having the fit value larger than 0.8 were retained (the maximum fit value was scaled to be 1.0), which were used in the following screening of molecular docking.
Molecular docking can generate the optimal binding mode of a ligand with a given receptor on the basis of docking scores[27], which can filter the false positives and improve the hit rate. The docking protein was prepared using the Prepare Protein program in DS 3.0. The initial water molecules and other crystallographic additives were removed, and the incomplete side-chain backbone atoms in the protein chains were fixed. The enumeration of hydrogen and protonation state was corrected according to the specified physiological PH. The binding region was defined to encompass the protein atoms within a sphere of 10 Å radius centered on a native ligand, using Receptor-Ligand Interactions protocol. This native ligand was then extracted from the crystal complex to perform molecular docking. The top 10 hits with high docking scores were kept for further analysis.
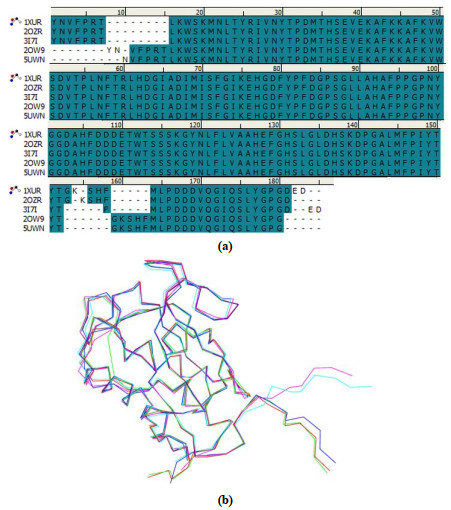
In order to build the multiple receptor-ligand based pharma- cophore model of MMP-13 inhibitors, we first compared the structures of five MMP-13 receptors in the above five experimental MMP-13-ligand complexes, 1XUR, 2OW9, 2OZR, 3I7I, and 5UWN. From Fig. 2(a) and (b), we can see that for these five MMP-13 receptors, their amino acid sequences are very similar (95.3%), and their structures are also very similar (96.2%).
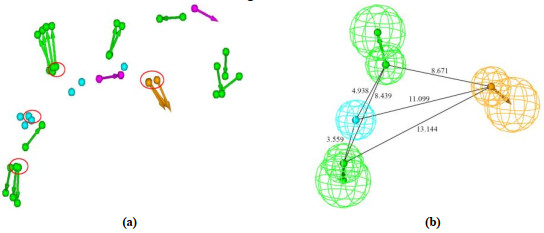
Based on the above five experimental MMP-13-ligand complexes, the corresponding five pharmacophore hypotheses were first generated, respectively. Then chemical features for these five pharmacophore hypotheses were superimposed, as shown in Fig. 3(a), based on the excellent overlap in Fig. 2. Next, the hierarchical clustering was carried out for these pharmacophore features to cover their common features using cluster protocols. The best cluster centers for chemical features were highlighted using the red circles, as shown in Fig. 3(a), which was used to build the final pharmacophore model. Finally, Fig. 3(b) shows the built pharmacophore model of MMP-13 inhibitors with distance constrains, which contains the four chemical features, i.e., two hydrogen bond acceptors (HBA), one hydrophobic (HY) feature, and one ring aromatic (RA) feature.
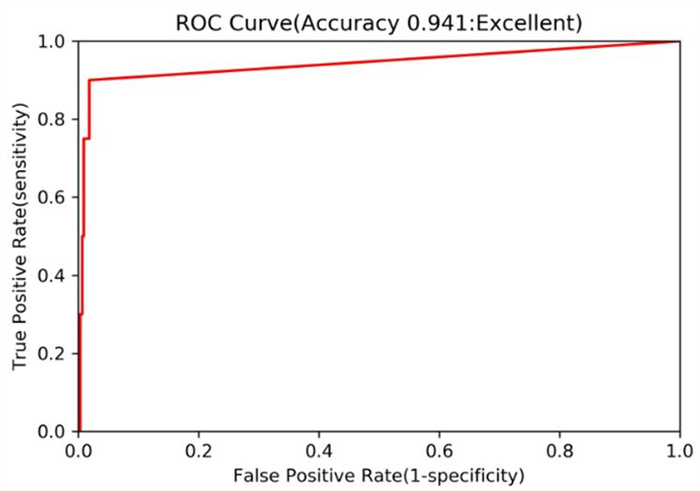
A valid pharmacophore model should be predictive to external data sets. Here, we employed a decoy set to validate the reliability for the built pharmacophore model. Table 1 shows the results of screening test from the decoy set. In Table 1, the decoy set, consisting of 1220 compounds (D) downloaded from Binding Database, was manually constructed for the purpose of validating the pharmacophore model. In this decoy set, there are 20 known active non-zinc-chelating inhibitors of MMP-13 (A) and the 1200 inactive compounds. After screening from the decoy set based on the built pharmacophore model, the 40 compounds (Ht) were recovered as the total hits. In these 40 compounds, the 18 compounds (Ha) are the active hits of the known MMP-13 inhibitors; hence, the percentage (%A) for active hits is 45, whereas, in the total 20 known MMP-13 inhibitors, the percentage (%Y) for these active hits is 90, indicating the much higher active hits. Our calculated enrichment factor (EF) is 27.45, which suggests the better ability of pharmacophore model to search the active compounds from a database. Moreover, the Güner-Henry (GH) score was calculated to be 0.652. Generally, the GH score of 0.6~1 indicates a very good pharmacophore model. Fig. 4 further shows the performance of receiver operating characteristic (ROC) regarding the pharmacophore model to screen the decoy set. The area under the ROC curve (AUC) was calculated to be 0.941. Such a high area indicates that the pharmacophore model has good screening ability. According to the above results, our built pharmacophore model was validated to be reliable.
 DownLoad:
CSV
DownLoad:
CSV
| Parameter | Decoy set |
| Total number of compounds in decoy set (D) | 1220 |
| Total number of actives in decoy set (A) | 20 |
| Total hits from decoy set (Ht) | 40 |
| Active hits for known MMP-13 inhibitors (Ha) | 18 |
| % yield of actives (%A) [(Ha/Ht)*100] | 45 |
| % ratio of actives (%Y) [(Ha/A)*100] | 90 |
| False negatives (A – Ha) | 2 |
| False positives (Ht – Ha) | 22 |
| Enrichment factor (EF) [(Ha*D)/(Ht*A)] | 27.45 |
| Güner-Henry (GH) score* | 0.652 |
| *GH = [Ha/(4Ht × A)](3A + Ht)[1–(Ht–Ha)/(D–A)] | |
To identify the novel selective MMP-13 inhibitors, the validated pharmacophore model of MMP-13 inhibitors was employed as a query to perform the database screening. Firstly, all the 61953 natural products in both AnalytiCon Discovery and InterBioScreen databases were filtered through Lipinski's rule of five to ensure the drug-like properties, where the 53648 drug-like compounds were obtained. Then, the pharmacophore model was employed to screen these drug-like compounds, and the 3126 compounds having a fit value range of 0.52~0.97 were retrieved. Finally, the 368 compounds with fit value > 0.8 were retained to increase the hit rate, and they were used in the following screening of molecular docking.
Our previous studies[28, 29] have shown that the CDOCKER docking program can successfully predict the experimental binding mode of a ligand with MMP-2 or MMP-9 receptor where the zinc-chelating MMP-2 or MMP-9 inhibitors were involved. Here, the five native ligands (Fig. 1) from the above five experimental MMP-13-ligand complexes (1XUR, 2OW9, 2OZR, 3I7I, and 5UWN) were docked back into their corresponding MMP-13 receptors to further examine the reliability of CDOCKER docking results where the non-zinc-chelating MMP-13 inhibitors were involved. After docking, we calculated the root-mean-square deviations (RMSD) relative to their corresponding experimental poses. The RMSD values involving the 1XUR, 2OW9, 2OZR, 3I7I, and 5UWN were calculated to be 0.5, 1.1, 0.2, 2.1 and 1.4, respectively, and the average RMSD and standard deviations were calculated to be only 1.1 and 0.8, respectively, which show that the CDOCKER program is reliable and can be employed to perform our screening of molecular docking.
According to the above results of database screening, the retained 368 compounds were used to further perform the screening of molecular docking to avoid the false positive. By using the CDOCKER docking program, all these 368 compounds were docked into the active sites of MMP-13 receptor involved in the above 2OZR MMP-13-ligand complex. We identified the selective non-zinc-chelating MMP-13 inhibitors based on the observations of following key docked poses. On one hand, the substituent groups of every natural product do not interact with the catalytic zinc ion of MMP-13 to screen the non-zinc-chelating MMP-13 inhibitors. On the other hand, every natural product should have a hydrophobic long chain group that can well map with the one hydrophobic (HY) feature of pharmacophore model displayed in Fig. 3(b). Such a HY feature will orient the natural product deep into the S1' pocket to increase the hydrophobic contacts in the tunnel region beneath the S1' specificity loop, which is able to produce the selective inhibition of natural product against MMP-13. Besides, the hydrogen bonds and the other intermolecular interactions, producing between the substituent groups of natural product and the amino acid residues in the S1' pocket of MMP-13, should have the important influence on strengthening the selective inhibitory ability of natural product against MMP-13. On the basis of the above observations of key docked poses, we found the four selective non-zinc-chelating MMP-13 inhibitors of natural products, including NP-015973, NP-000814, STOCK1N-24933, and STOCK1N-69443, and Fig. 5 reveals their molecular structures. Previous studies have shown that our screened natural products have some pharmacological properties. For example, NP-000814, isolated from the rhizomes of Curcuma kwangsiensis, can produce the inhibition against the NO production with IC50 value of 5.58 μM[30]. Besides, NP-015973, isolated from the bee pollen of Quercus mongolica, has the inhibitory ability against tyrosinase with IC50 value of 75.1 μM[31].
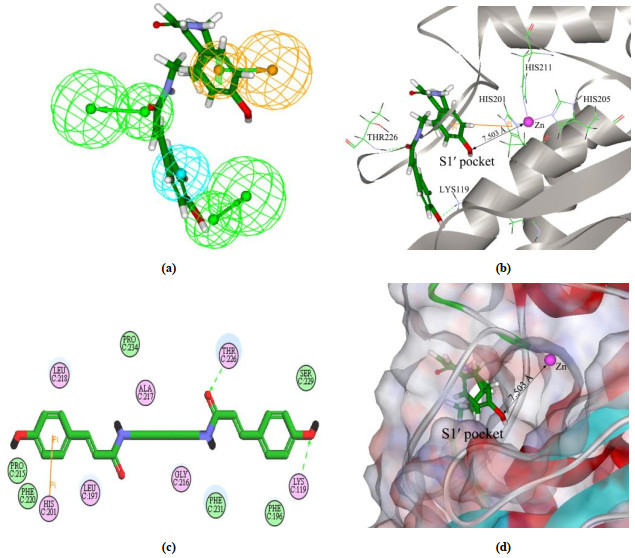
In order to investigate the binding modes, we used one of the four screened inhibitors displayed in Fig. 5, NP-015973, as an example to elucidate the interaction ways between NP-015973 and MMP-13 active sites. Fig. 6 shows the results of both pharmacophore mapping and molecular docking concerning NP-015973. As seen in Fig. 6(a), NP-015973 can well map with all the four chemical features of pharmacophore model. Particularly, a hydrophobic long chain group in the molecular structure of NP-015973 can well map with the hydrophobic (HY) feature of pharmacophore model, which is able to orient the ligand NP-015973 deep into the S1' pocket of MMP-13 to produce the selective inhibition against MMP-13. In Fig. 6(b), (c), and(d), the three different representations are used to display the binding mode of NP-015973 with MMP-13 active sites. We see that the shortest distance between the substituent groups of NP-015973 and the catalytic zinc ion of MMP-13 is 7.503 Å, so NP-015973 does not interact with the catalytic zinc ion of MMP-13, as shown in Fig. 6(b) and (d). Therefore, NP-015973 is non-zinc-chelating against MMP-13. Previous studies have shown that most of the MMP inhibitors are zinc-chelating, which should be the important reason why many of them lackelective inhibition against MMP-13[7, 8].Fig. 6(b), (c), and(d) further reveals that the whole NP-015973 molecule has almost entered the S1' pocket of MMP-13 to produce the following intermolecular interactions. One interaction way is that there are two hydrogen bonds producing between the substituent groups (carbonyl and hydroxyl) of NP-015973 and the amino acid residues (Thr226 and Lys119) of the S1' pocket in MMP-13. Another interaction way is that a special π-π stacking forms between the phenylic ring of NP-015973 and the imidazole ring of His201 in MMP-13. In conclusion, the above results show that NP-015973 can well map with the built pharmacophore model, and although the ligand NP-015973 is non-zinc-chelating against MMP-13, it can better bind with the S1' pocket of MMP-13. Therefore, NP-015973 should be the selective non-zinc-chelating MMP-13 inhibitor of natural product.
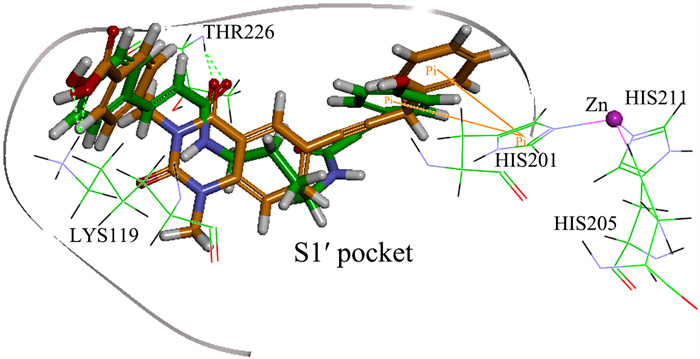
To examine the reliability of screened NP-015973 discussed above, Fig. 7 further displays the superposition of the two complex structures regarding the MMP-13 receptor binding with both our screened ligand NP-015973 and the most active ligand GG12003 (IC50: 0.67 nM) among the five experimental crystal structures of MMP-13-ligand complexes, as shown in Fig. 1. From Fig. 7, we see that the substituent groups of both ligands NP-015973 and GG12003 do not interact with the catalytic zinc ion of MMP-13 receptor, so they are non-zinc-chelating against MMP-13. Interestingly, it is found that the binding modes of these two ligands with the MMP-13 receptor are very similar, which is described as follows: One carbonyl group and one hydroxyl group in each of these two ligands are able to form the hydrogen bonds with the amino acid residues Thr226 and Lys119 of the S1' pocket in MMP-13 receptor, respectively. Besides, one phenylic ring in each of these two ligands can form π-π stacking with the imidazole ring of His201 in MMP-13 receptor. The above obvious similarity of two binding modes, involving the MMP-13 receptor binding with both our screened NP-015973 and the experimental most active inhibitor GG12003, shows that our modeling results are in good agreement with the relevant experimental results, indicating that our screened NP-015973 should be the selective non-zinc-chelating inhibitors of MMP-13.
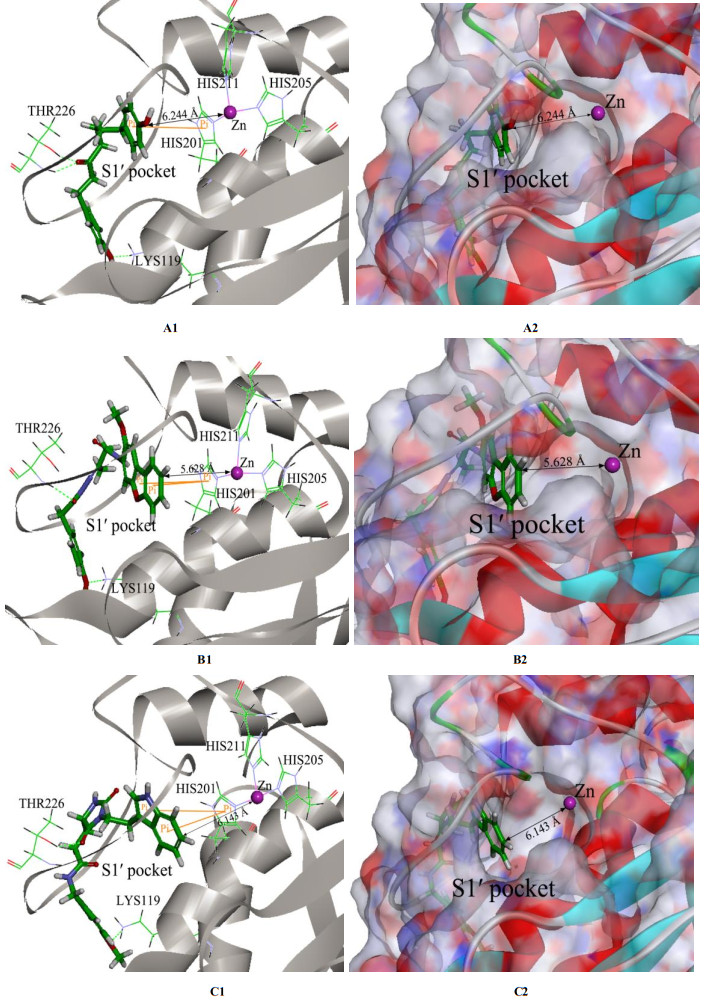
Finally, we further inspect the relevant results of molecular docking for the other three screened natural products, NP- 000814, STOCK1N-24933, and STOCK1N-69443 displayed in Fig. 5, with MMP-13 receptor, and all their binding modes are shown in Fig. 8. From Fig. 8, we see that like NP-015973 discussed above, each natural product does not interact with the catalytic zinc ion of MMP-13 because of the farther distance between them, so these three natural products are non-zinc-chelating against MMP-13. Besides, the whole molecule of each natural product almost enters the S1' pocket of MMP-13 to produce the interaction ways involving the two or three hydrogen bonds and the π-π stacking, which are also very similar to the interaction ways of MMP-13 with the experimental most active inhibitor GG12003 as shown in Fig. 7, and they all involve the interactions of a ligand with the three amino acid residues Thr226, Lys119, and His201 of MMP-13 receptor, showing that our modeling results concerning these three natural products are also in good agreement with the relevant experimental results. The above results indicate that these three natural products should also be the selective non-zinc-chelating inhibitors of MMP-13, and they may be used as the lead compounds in the future studies of structural modifications.
In this study, the experimental crystal structures of multiple receptor-ligand complexes have been used to build the pharmacophore model of MMP-13 inhibitors. By using the decoy set, we validate that the pharmacophore model is reliable. It is found that the pharmacophore model contains the four chemical features, i.e., two HBA, one HY, and one RA features. Particularly, the HY feature is found to orient the MMP-13 inhibitors deep into the S1' pocket of MMP-13 to produce selective inhibition. Using the experimental crystal structures of five MMP-13-ligand complexes, we validate that the CDOCKER docking program is reliable and can be employed to perform the screening of molecular docking. According to the screening of pharmacophore model and subsequent molecular docking, the four selective non-zinc- chelating MMP-13 inhibitors of natural products are identified, which includes NP-015973, NP-000814, STOCK1N-24933 and STOCK1N-69443. The further study shows that the binding modes of MMP-13 with our screened four natural products are very similar to the reported experimental binding mode of MMP-13 with most active inhibitor of GG12003, and they all involve the interactions of a ligand with the three amino acid residues Thr226, Lys119, and His201 of MMP-13 receptor. The good agreement of our screening results with the relevant experimental results strongly supports our screened MMP-13 inhibitors of natural products. These screened natural products may be used as the lead compounds of MMP-13 inhibitors in the future studies of structural modifications.
Stamenkovic, I. Extracellular matrix remodelling: the role of matrix metalloproteinases. J. Pathol. 2003, 200, 448−464. doi: 10.1002/path.1400
Murphy, G.; Knauper, V.; Atkinson, S.; Butler, G.; English, W.; Hutton, M. Matrix metalloproteinases in arthritic disease. Arthritis Res. 2002, 4, S39−S49. doi: 10.1186/ar572
Conrozier, T.; Ferrand, F.; Poole, A. R.; Verret, C. Differences in biomarkers of type II collagen in atrophic and hypertrophic osteoarthritis of the hip: implications for the differing pathobiologies. Osteoarthr Cartilage. 2007, 15, 462−467. doi: 10.1016/j.joca.2006.09.002
Buckwalter, J. A.; Martin, J. A. Osteoarthritis. Adv. Drug Delivery Rev. 2006, 58, 150−167. doi: 10.1016/j.addr.2006.01.006
Neuhold, L. A.; Killar, L.; Zhao, W.; Sung, M. A.; Warner, L.; Kulik, J.; Turner, J. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Invest. 2001, 107, 35−44. doi: 10.1172/JCI10564
Stickens, D.; Behonick, D. J.; Ortega, N.; Heyer, B.; Hartenstein, B.; Yu, Y.; Fosang, A. J.; Angel, P.; Werb, Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 2004, 131, 5883−5895. doi: 10.1242/dev.01461
Tallant, C.; Marrero, A. Matrix metalloproteinases: fold and function of their catalytic domains. Biochim. Biophys. Acta 2010, 1803, 20−28. doi: 10.1016/j.bbamcr.2009.04.003
Renkiewicz, R.; Qiu, L.; Lesch, C.; Sun, X.; Devalaraja, R.; Cody, T.; Kaldjian, E.; Welgus, H.; Baragi, V. Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 2003, 48, 1742−1749. doi: 10.1002/art.11030
Fabre, B.; Ramos, A. Targeting matrix metalloproteinases: exploring the dynamics of the S1' pocket in the design of selective, small molecule inhibitors. J. Med. Chem. 2014, 57, 10205−10219. doi: 10.1021/jm500505f
Engel, C. K.; Pirard, B.; Schimanski, S.; Kirsch, R.; Habermann, J.; Klingler, O.; Schlotte, V.; Weithmann, K. U.; Wendt, K. U. Structural basis for the highly selective inhibition of MMP-13. Chem. Biol. 2014, 12, 181−189.
Johnson, A. R.; Pavlovsky, A. G.; Ortwine, D. F.; Prior, F.; Man, C. F.; Bornemeier, D. A.; Banotai, C. A. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J. Biol. Chem. 2007, 282, 27781−27791. doi: 10.1074/jbc.M703286200
Heim-Riether, A.; Taylor, S. J.; Liang, S.; Gao, D. A. Improving potency and selectivity of a new class of non-Zn-chelating MMP-13 inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5321−5324. doi: 10.1016/j.bmcl.2009.07.151
Choi, J. Y.; Fuerst, R.; Knapinska, A. M. Structure-based design and synthesis of potent and selective matrix metalloproteinase 13 inhibitors. J. Med. Chem. 2017, 60, 5816−5825. doi: 10.1021/acs.jmedchem.7b00514
Gao, D. A.; Xiong, Z.; Heim-Riether, A.; Amodeo, L.; August, E. M.; Cao, X.; Ciccarelli, L. SAR studies of non-zinc-chelating MMP-13 inhibitors: improving selectivity and metabolic stability. Bioorg. Med. Chem. Lett. 2010, 20, 5039−5043. doi: 10.1016/j.bmcl.2010.07.036
Gege, C.; Bao, B.; Bluhm, H.; Boer, J.; Gallagher, B. M.; Korniski, B.; Powers, T. S.; Steeneck, C. Discovery and evaluation of a non-zn chelating, selective matrix metalloproteinase 13 (MMP-13) inhibitor for potential intra-articular treatment of osteoarthritis. J. Med. Chem. 2012, 55, 709−716. doi: 10.1021/jm201152u
Li, J. J.; Nahra, J.; Johnson, A. R.; Bunker, A.; O'Brien, P.; Yue, W. S.; Ortwine, D. F.; Man, C. F.; Baragi, V. Quinazolinones and pyrido [3, 4-d] pyrimidin-4-ones as orally active and specific matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. J. Med. Chem. 2008, 51, 835−841. doi: 10.1021/jm701274v
Taylor, S. J.; Abeywardane, A.; Liang, S.; Muegge, I.; Padyana, A.; Xiong, Z.; Hill-Drzewi, M.; Farmer, B. Fragment based discovery of indole inhibitors of matrix metalloproteinase-13. J. Med. Chem. 2011, 54, 8174−8187. doi: 10.1021/jm201129m
Savi, C. D.; Morley, A. D.; Ting, A.; Nash, I.; Karabelas, K.; Wood, C. M.; James, M.; Norris, S. J.; Karoutchi, G.; Rankine, N. Selective non zinc binding inhibitors of MMP13. Bioorg. Med. Chem. Lett. 2011, 21, 4215−4219. doi: 10.1016/j.bmcl.2011.05.075
Spicer, T. P.; Jiang, J.; Taylor, A. B.; Choi, J. Y.; Hart, P. J.; Roush, W. R.; Field, G. B.; Hodder, P. S.; Minond, D. Characterization of selective exosite-binding inhibitors of matrixmetalloproteinase 13 that prevent articular cartilage degradation in vitro. J. Med. Chem. 2014, 57, 9598−9611. doi: 10.1021/jm501284e
Cragg, G. M.; Grothaus, P. G.; Newman, D. J. New horizons for old drugs and drug leads. J. Nat. Prod. 2014, 77, 703−723. doi: 10.1021/np5000796
Cragg, G. M.; Grothaus, P. G.; Newman, D. Impact of natural products on developing new anti-cancer agents. J. Chem. Rev. 2009, 109, 3012−3043. doi: 10.1021/cr900019j
Yang, S. Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discovery Today 2010, 15, 444−450. doi: 10.1016/j.drudis.2010.03.013
Gagnon, J. K.; Law, S. M.; Brooks, C. L. Flexible CDOCKER: development and application of a pseudo-explicit structure-based docking method within CHARMM. J. Comput. Chem. 2016, 37, 753−762. doi: 10.1002/jcc.24259
Discovery Studio, Version 3.0; Accelrys Inc: San Diego 2010.
Hamza, A.; Wei, N. N.; Zhan, C. G. Ligand-based virtual screening approach using a new scoring function. J. Chem. Inf. Model 2012, 52, 963−974. doi: 10.1021/ci200617d
Triballeau, N.; Acher, F.; Brabet, I. Virtual screening workflow development guided by the "receiver operating characteristic" curve approach, application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534−2547. doi: 10.1021/jm049092j
Hein, M.; Zilian, D. Docking compared to 3D pharmacophores: the scoring function challenge. Drug Discovery Today: Technol. 2011, 7, e229−e236.
Wang, Y. J.; Yang, L. M.; Hou, J. Y.; Zou, Q.; Gao, Q.; Yao, W. H.; Yao, Q. Z.; Zhang, J. Hierarchical virtual screening of the dual MMP-2/HDAC-6 inhibitors from natural products based on pharmacophore models and molecular docking. J. Biomol. Struct. Dyn. 2019, 37, 649−670. doi: 10.1080/07391102.2018.1434833
Hou, J. Y.; Zou, Q.; Wang, Y. J.; Gao, Q.; Yao, W. H.; Yao, Q. Z.; Zhang, J. Screening for the selective inhibitors of MMP-9 from natural products based on pharmacophore modeling and molecular docking in combination with bioassay experiment, hybrid QM/MM calculation, and MD simulation. J. Biomol. Struct. Dyn. 2019, 37, 3135−3149. doi: 10.1080/07391102.2018.1509019
Li, J.; Zhao, F.; Li, M. Z.; Chen, L. X.; Qiu, F. Diarylheptanoids from the Rhizomes of Curcuma kwangsiensis. J. Nat. Prod. 2010, 73, 1667−1671. doi: 10.1021/np100392m
Kim, S. B.; Liu, Q.; Ahn, J. H.; Jo, Y. H.; Turk, A.; Hong, I. P.; Han, S. M.; Hwang, Y. B.; Lee, M. K. Polyamine derivatives from the bee pollen of Quercus mongolica with tyrosinase inhibitory activity. Bioorg. Chem. 2018, 81, 127−133. doi: 10.1016/j.bioorg.2018.08.014

Figure 1 Molecular structures of five ligands involved in the five experimental MMP-13-ligand complexes and the IC50 values

Figure 2 (a) Amino acid sequences and (b) structural superimposition of the five MMP-13 receptors in the five experimental MMP-13-ligand complexes. Red, blue, cyan, purple, and green represent the five MMP-13 receptors in the five MMP-13-ligand complexes 1XUR, 2OZR, 2OW9, 5UWN, and 3I7I, respectively

Figure 3 (a) Superimposition of chemical features for the five pharmacophore hypotheses that were produced by the five experimental crystal structures of MMP-13-ligand complexes respectively. The red circles denote the best cluster centers of feature groups. (b) The built pharmacophore model of MMP-13 inhibitors with distance constrains, generated by the best cluster centers that were denoted using the red circles as shown in (a), where green, cyan, and orange denote hydrogen bond acceptor (HBA), hydrophobic (HY) feature, and ring aromatic (RA) feature, respectively

Figure 4 Performance of receiver operating characteristic (ROC) obtained from the screening test of decoy set using the pharmacophore model

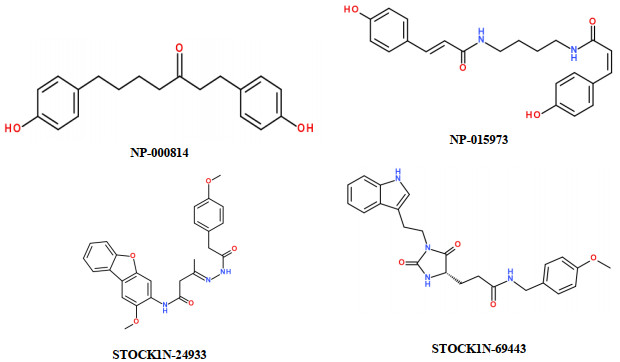
Figure 5 Molecular structures for the screened four selective non-zinc-chelating MMP-13 inhibitors of natural products

Figure 6 (a) Screened NP-015973 mapping with the pharmacophore model. The meaning of color for each pharmacophore feature can be seen in Fig. 3. (b), (c), and (d) represent the 3D, 2D and surface structures of binding mode for the screened NP-015973 with MMP-13 receptor, respectively, obtained from molecular docking. The black line with two arrows denotes the distance between Zn and O. The hydrogen bond and π-π stacking are exhibited by green broken line and orange line, respectively

Figure 7 Superposition of the two complex structures regarding the MMP-13 receptor binding with both our screened ligand NP-015973 and the experimental most active ligand GG12003 shown in Fig. 1. The carbonic atoms in ligands NP-015973 and GG12003 are displayed by green and orange, respectively. The hydrogen bond and π-π stacking are exhibited by green broken line and orange line, respectively

Figure 8 3D structures and surface structures of binding modes for the other three screened natural products with the MMP-13 receptor obtained from molecular docking, which are denoted by A1 and A2 for NP-000814, respectively, B1 and B2 for STOCK1N-24933, respectively, and C1 and C2 for STOCK1N-69443, respectively. The black line with two arrows denotes the distance between Zn and O. The hydrogen bond and π-π stacking are exhibited by green broken line and orange line, respectively
Table 1. Fig. 1. Molecular structures of five ligands involved in the five experimental MMP-13-ligand complexes and the IC50 values Table 1. Results of Screening Test from the Decoy Set
| Parameter | Decoy set |
| Total number of compounds in decoy set (D) | 1220 |
| Total number of actives in decoy set (A) | 20 |
| Total hits from decoy set (Ht) | 40 |
| Active hits for known MMP-13 inhibitors (Ha) | 18 |
| % yield of actives (%A) [(Ha/Ht)*100] | 45 |
| % ratio of actives (%Y) [(Ha/A)*100] | 90 |
| False negatives (A – Ha) | 2 |
| False positives (Ht – Ha) | 22 |
| Enrichment factor (EF) [(Ha*D)/(Ht*A)] | 27.45 |
| Güner-Henry (GH) score* | 0.652 |
| *GH = [Ha/(4Ht × A)](3A + Ht)[1–(Ht–Ha)/(D–A)] | |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们