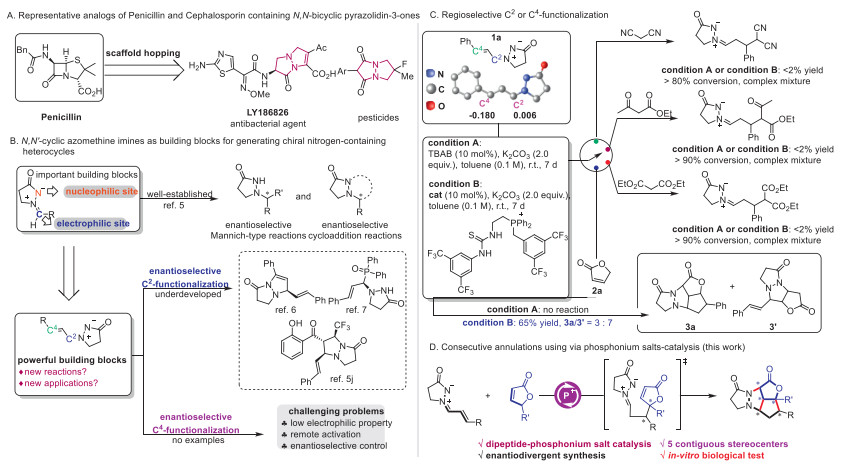
Scheme 1.
Applications of N,N-bicyclic pyrazolidine-3-ones and new reactivities of N,N′-cyclic azomethine imines for generating chiral polyheterocyclic compounds. TBAB: tetrabutylammonium bromide.
Enantioselective regulation to coronal polyheterocyclic compounds via phosphonium salt-catalyzed cycloadditions of azomethine imines with γ-butenolides
Jun Liu , Zhaoyu Feng , Renming Pan , Xiaolong Yu , Meijuan Zhou , Gang Zhao , Hongyu Wang

β-Lactam antibiotics, such as penicillin and cephalosporin, represent one of the most important classes of antibacterial agents globally [1,2]. Despite the discovery and clinical use of numerous β-lactams over the past decades, the search for better drugs and solutions to antibiotic resistance continues [3–5]. So medicinal chemists have focused on scaffold hopping strategies through synthetic modifications of the β-lactam ring in this pursuit [6–8]. To date, various pyrazolidinones have been developed due to their distinct bioactivity and excellent antibacterial activity [9–11], which function as β-lactam analogs and show significant potential as effective antibacterial agents (Scheme 1A).
To date, the enantioselective generation of pyrazolidinones has garnered significant interest from chemists [12–14]. Consequently, N,N'-cyclic azomethine imines have emerged as important building blocks for this research, greatly accelerating the development of N,N-bicyclic pyrazolidine-3-one synthesis. This has facilitated the construction of diverse structures and molecular libraries through asymmetric Mannich-type reactions [15–17] and cycloaddition reactions catalyzed [18–23] by both metal catalysts and organocatalysts (Scheme 1B, top). Of note, alkenyl azomethine imines derived from α,β-unsaturated aldehydes [24,25] with multiple reactive sites have also been explored in recent years. For example, Kobayashi and co-workers first reported an enantioselective synthesis using alkenyl azomethine imine and terminal alkyne catalyzed by a copper complex to generate chiral N,N-bicyclic pyrazolidinone [26]. Wang and co-workers also achieved an enantioselective phosphination of alkenyl azomethine imine via an asymmetric Mannich-type reaction using chiral squaramide [27]. Recently, Chang and co-workers realized an enantioselective synthesis of chiral N,N-bicyclic pyrazolidinone through a [3 + 2] cycloaddition of β-trifluoromethyl α,β-unsaturated ketone with alkenyl azomethine imine [28]. Although some examples of enantioselective transformations utilizing alkenyl azomethine imines have been reported, they generally exhibit similar reactivity to azomethine imines derived from aryl aldehydes, with only C2-functionalization being achieved (Scheme 1B, below). Exploring new reactivities of alkenyl azomethine imines is highly desirable due to their easy availability and versatile alkenyl substituents, potentially paving the way for the generation of diverse chiral N,N-bicyclic pyrazolidine-3-ones and related compounds. Consequently, our attention has been drawn to the enantioselective C4-functionalization of alkenyl azomethine imines, which remains to be realized.
Initially, we examined malononitrile, ethyl acetoacetate, and diethyl malonate as nucleophiles with alkenyl azomethine imine 1a using K2CO3 and TBAB in toluene (Scheme 1C). However, no 1,4-conjugate addition products were observed by GC–MS analysis, even when thiourea-phosphonium salt, a powerful phase-transfer catalyst in our previous works [29–31], was utilized in these reactions. NBO charge analysis shows that the C2-site (0.006 e) of 1a is significantly more electrophilic than the C4-site (-0.180 e), which explains why C2-functionalization of 1a is more favorable, making the 1,4-conjugate addition of alkenyl azomethine imine a challenging problem [32,33]. As we all know, azomethine imines are good 1,3-dipoles in organic synthesis [34], which inspired us that 1,3-dipolar cycloaddition reactions following the 1,4-conjugate addition would solve the above problems. Thus, furan-2(5H)-one, which is an important γ-butenolide bearing a pro-nucleophilic site and an alkenyl group [35–38], was then examined with alkenyl azomethine imine under proton-transfer conditions (Scheme 1C, below). To our delight, an interesting coronal polyheterocyclic compound (3a) was obtained via a formal [5 + 3] cycloaddition process, even though with a low yield, when thiourea-phosphonium salt as the phase-transfer catalyst was utilized, but no product was generated without the quaternary phosphonium salt. Additionally, the product (3′) via [3 + 2] cycloaddition pathway was generated with higher yield in comparison with 3a, which also suggested that C2-functionalization of alkenyl azomethine imines was favorable, and C4-functionalization was much more challenging. Given the importance of the phosphonium salt in accelerating the cycloaddition reactions, we thought that optimizing the reaction conditions, including the phosphonium salts, solvents, bases, could selectively realize the C4-functionalization of alkenyl azomethine imines to generate 3a. Herein, an enantioselective version was carried out in this work to synthesize enantioenriched polyheterocyclic compounds, further evaluation of their biological activity and DFT calculations were also performed to illustrate their applications and the reaction mechanism.
Enantioselective cycloaddition reaction between alkenyl azomethine imines 1a and furan-2(5H)-one 2a was selected as the model reaction. Given the powerful and sustainable approach for asymmetric synthesis via dipeptide-phosphonium salts, which have been extensively explored by Zhao, Wu, and Wang [29–31,39–41], the chiral quaternary phosphonium salts were then carefully examined in the reaction. As shown in Table 1, cat-1 derived from L-tert-leucine and L-phenylglycine was first explored to screen the solvents, such as toluene, TBME, CH2Cl2, and anisole (Table 1, entries 1–4). The ee value of 3a could be improved to 70% when anisole was used as the solvent. Optimization on the effects of bases in the reaction revealed that K2HPO4 was the best choice (Table 1, entries 4–7), which could improve the ratio of 3a/3′ to > 20:1, and encouraged us to further screen the phosphonium salts. Then, exploration of the catalysts' structure was continued to be performed via tuning the chiral amino acids (Table 1, entries 8–11). When L-tert-leucine and L-tert-leucine derived dipeptide-phosphonium salt cat-5 was used in this reaction (Table 1, entry 11), the ee value could be improved to 93%, and 94% yield was obtained simultaneously, so the bulky steric effect of the catalysts is much more favorable for increasing enantioselectivities. The absolute configuration of 3a was then determined by X-ray crystallography analysis (CCDC: 2156978). Of note, cat-6 can reverse the absolute configuration of the corresponding product with a high ee value (Table 1, entry 12, 3a′), which just needs to modify hydrogen binding groups [31]. The absolute configurations of 3a′ was also determined by X-ray crystallography analysis (CCDC: 2350960). Meanwhile, lowering the catalyst loading of cat-6 to 3 mol% was not detrimental to the yield and enantioselectivity (Table 1, entry 13). So it provides an enantiodivergent and simple approach accessing to coronal polyheterocyclic compounds.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||||
| Entry | Cat. | Base | Solvent | 3a/3′ | Yield (%)b | ee of 3a (%)c |
| 1d | cat-1 | K2CO3 | Toluene | 4/6 | 64 | 50 |
| 2d | cat-1 | K2CO3 | TBME | 6/4 | 57 | 48 |
| 3 | cat-1 | K2CO3 | CH2Cl2 | 8/2 | 64 | 38 |
| 4 | cat-1 | K2CO3 | Anisole | 6/4 | 75 | 70 |
| 5 | cat-1 | K2HPO4 | Anisole | > 20/1 | 93 | 80 |
| 6 | cat-1 | K3PO4 | Anisole | 3/7 | 55 | 71 |
| 7 | cat-1 | Cs2CO3 | Anisole | 2/8 | 46 | 72 |
| 8 | cat-2 | K2HPO4 | Anisole | > 20/1 | 95 | 70 |
| 9 | cat-3 | K2HPO4 | Anisole | > 20/1 | 79 | 79 |
| 10 | cat-4 | K2HPO4 | Anisole | > 20/1 | 60 | 7 |
| 11 | cat-5 | K2HPO4 | Anisole | > 20/1 | 94 | 93 |
| 12 | cat-6 | K2HPO4 | Anisole | > 20/1 | 95 | -87 |
| 13e | cat-6 | K2HPO4 | Anisole | > 20/1 | 93 | -87 |
| a Unless otherwise specified, the reaction was carried out with 1a (0.10 mmol), 2a (0.15 mmol), catalyst (5 mol%), and base (0.2 mmol) in anisole (1.0 mL) at room temperature for 48 h. b Isolated yields of product. c Determined by chiral HPLC analysis. d The reaction time is 7 days. e With 3.0 mol% cat-6. dr of 3a were > 20:1. |
||||||
With the optimized conditions in hand, the substrate scope continued to be explored through the modification of electronic effects and steric effects of the substrates. As shown in Scheme 2 (top), para-substituents on the aryl ring of 1 were all well tolerated using cat-5 no matter electron-donating or electron-withdrawing substituents such as methoxy, nitro, amino, and fluorine (3b-3h). In addition, enantiodivergent generation of the corresponding products was also realized when cat-6 was used instead of cat-5, and high yields and excellent enantioselectivities were obtained (3b′-3h′). Then, meta- and ortho-substituents on the aryl rings were subsequently investigated under the best conditions, which showed that all could give high yield and ee (3i-3l′) even though moderate enantioselectivity was observed for 3k. In addition, naphthyl and furyl substituents could be also well tolerated in this reaction. When benzo[b]thiophene was introduced into the aryl ring of 1, the enantioselectivity was reduced to 72% ee (3o), while benzofuran was better suitable for this reaction with 81% ee (3p). Meanwhile, further exploration of the substituent effect showed that the cyclohexyl group was also tolerated in this catalytic system, and 94% yield and 95% ee of the corresponding product (3q) were furnished with 10:1 dr. The opposite absolute configuration 3p′ was also synthesized with 95% yield, 85% ee, and 20:1 dr through the utility of cat-6. Then, 5-methylfuran-2(5H)-one was subsequently examined by this method, 3q and 3q′ could be successfully generated with 95% ee and 85% ee respectively.
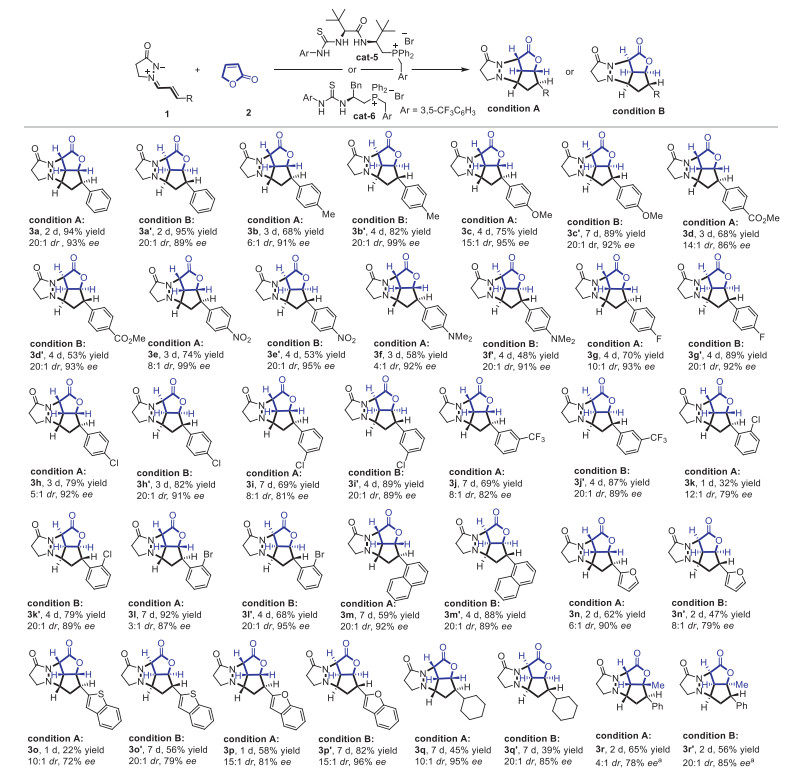
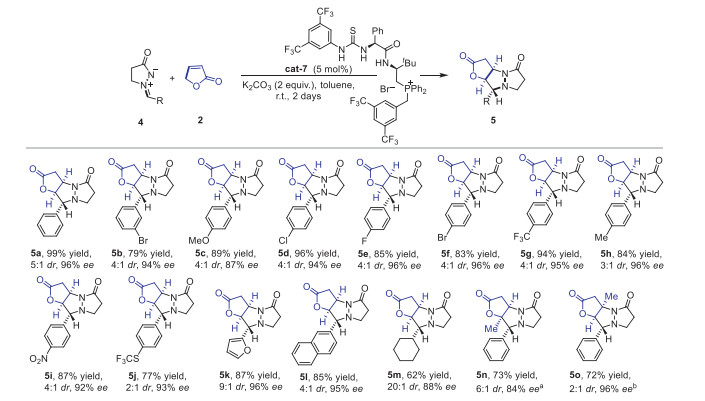
To expand the applications of the method, further investigations on enantioselective [3 + 2] cycloaddition between azomethine imines and furan-2(5H)-one was continued catalyzed by dipeptide-phosphonium salt (cat-7) (see Supporting information for optimizations on reaction conditions). As shown in Scheme 3, steric effects, electronic effects, and functional group compatibility on the aryl ring of 2 were all screened under the optimized conditions (5b-5j) such as halides, nitro, trifluoromethylthio. All the corresponding products were successfully obtained with excellent enantioselectivities (up to 96% ee) and high yields (up to 99% yield). Moreover, heterocyclic, polycyclic aromatic hydrocarbon, and alkyl groups were further examined, such as furyl, naphthalenyl, cyclohexyl, the desired products were all generated with high ee values and yields (88%-96% ee; 62%-99% yield; 5c-5m). In addition, 5-methylfuran-2(3H)-one and 4-methylfuran-2(5H)-one could react with 4a to give the corresponding product 5n and 5o respectively, and enantio-enriched quaternary carbon centers were successfully generated. The absolute configuration of 5o was then determined by X-ray crystallography analysis, and the other products were assigned by analogy (CCDC: 2156979). All the results of enantioselective [5 + 3] and [3 + 2] cycloaddition reactions demonstrate that alkenyl azomethine imines derived from α,β-unsaturated aldehydes possess unique and interesting properties, which can be applied for the generation of novel structural molecules.
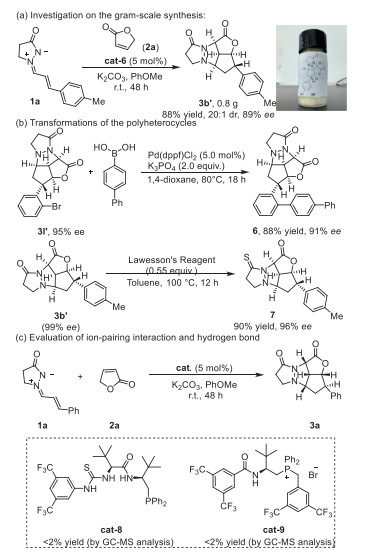
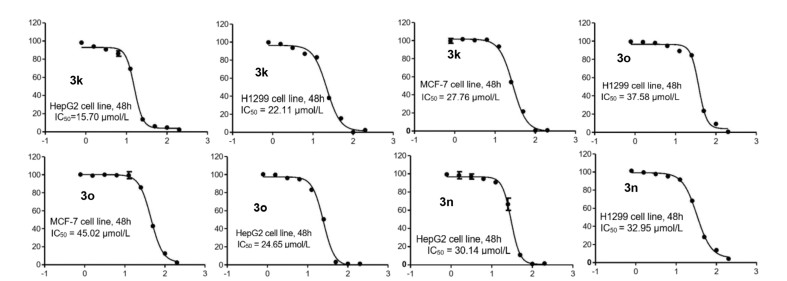
To illustrate the applications of the method, the gram-scale synthesis of 3b′ was then performed without a notable diminution in yield and enantioselectivity (Scheme 4a). Moreover, a Suzuki-Miyaura cross-coupling reaction using 3l′ and [1,1′-biphenyl]-4-ylboronic acid was performed to illustrate the further transformation of the polyheterocyclic compounds, and the corresponding product 6 was successfully obtained with high yields and the retained enantioselectivity (Scheme 4b). In addition, the amide group of 3b′ could be easily transformed into the thioamide (7) using Lawesson's reagent with high yield. To get insights into the catalytic mechanism of the phosphonium salt, further investigations on the rule of the quaternary phosphonium center and hydrogen bonds via the control experiments were also carried out subsequently. As shown in Scheme 4c, organophosphine cat-8 without phosphonium center and cat-9 with a single hydrogen bond all failed to realize the cycloaddition reaction, which herein indicated that both the dipeptide-phosphonium salt with thiourea moiety and the phosphonium center of the bifunctional phosphonium salts play crucial role in achieving the enantioselective [5 + 3] cycloaddition. With these chiral polyheterocyclic compounds in the hands, further evaluations of the biological activities in cells were continued to perform (Scheme 5). Then, the cytotoxic effects of some selected coronal polyheterocyclic compounds against a panel of cancer cell lines including human hepatoma HepG2 cells, H1299 lung cancer cells, and MCF-7 human breast cancer cells, were screened by cell counting kit-8 (CCK-8) assay. To our delight, 3k not only exhibits noticeable cytotoxicity against human hepatoma HepG2 cells (IC50 = 15.7 µmol/L), but also shows high cytotoxicity against H1299 lung cancer cells (IC50 = 22.11 µmol/L), and MCF-7 human breast cancer cells (IC50 = 27.76 µmol/L). 3o and 3n also exhibit cytotoxicity against these cells. Notably, the enantiomers of 3k, 3o, and 3n do not show cytotoxicity against cancer cells, which demonstrates the crucial role of absolute configuration in bioactivity.
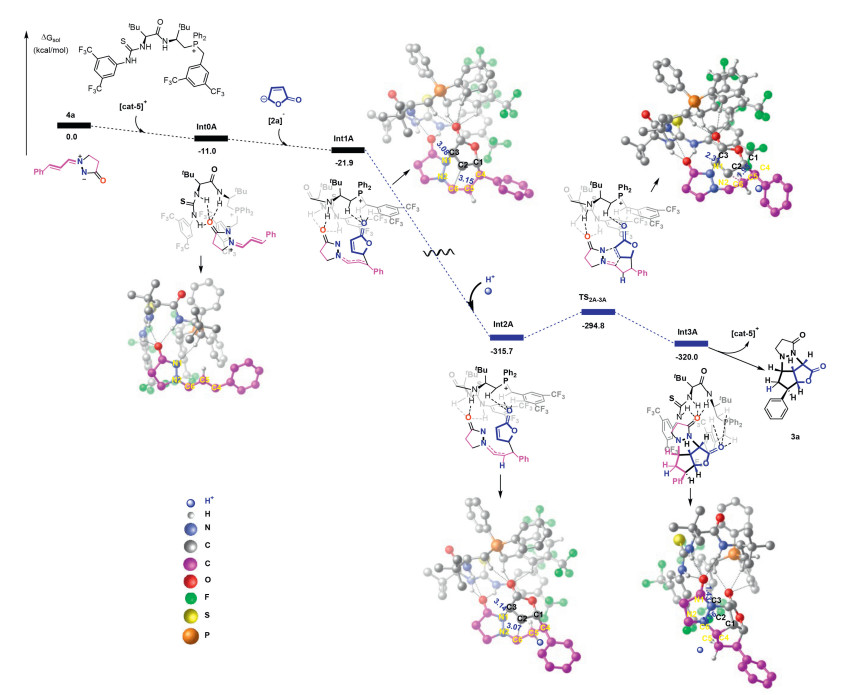
Based on the experimental studies, DFT calculations were carried out to illustrate the mechanisms behind these cycloaddition reactions. From the energy profile in Fig. 1, the alkenyl azomethine imines (4a) firstly is located at the active region of the catalyst of [cat-5]+ with the help of several hydrogen bonds formed between 4a and [cat-5]+ and the formation of the intermediate Int0A is exothermic by 11.0 kcal/mol. When the second reactant furan-2(5H)-one (2a) is involved in this reaction, 2a can easily lose a hydrogen proton to change into [2a]- with a negative charge under alkaline conditions in the experiments. Then the carbon atom C1 in the structure of [2a]- is bonded to the C4 atom of 4a to form the intermediate Int1A by an energy decrease of 10.9 kcal/mol based on the structure of Int0A. By analyzing the natural bond orbital (NBO) charges of Int1A in Fig. S7 (Supporting information), the carbon atom C5 in the structure of Int1A has the most negative charges of -0.367 |e| of the six carbon atoms from C1 to C6. Therefore, C5 atom tends to get one hydrogen proton under an acidic reaction condition. Here, for the mechanism of the enantioselective cycloaddition reaction of 4a and 2a catalyzed by cat-5, there are two possible pathways shown in Fig. 1 and Fig. S2 (Supporting information), respectively. If C5 atom gets one hydrogen proton to form another intermediate Int2A, the relative free energy of Int2A drops sharply. This is because the electrostatic interaction between the fragments is enhanced due to the charge transfer caused by the addition of hydrogen protons. Bycomparing the NBO charges of the corresponding atoms in the two structures of Int1A and Int2A in Fig. S7, the most obvious difference is the charges of C6 and C2. The net charge of C6 in Int2A is 0.137 |e| more positive than that in Int1A, however, the net charge of C2 in Int2A is 0.031 |e| more negative than that in Int1A. In addition, C6 and C2 in Int1A both bring negative charges, but the net charges of C6 and C2 are 0.124 |e| and -0.188 |e| in the structure of Int2A, respectively. From the view of NBO charge, Int2A has a greater advantage for the next cycloaddition reaction compared with Int1A, which is also confirmed in the energy barriers for the next step. In Fig. 1, based on the structure of Int2A, C2 and C3 atoms of [2a]- are, respectively bonded to C6 and N1 atoms of 4a to complete the next cycloaddition reaction by overcoming the energy barrier of 20.9 kcal/mol. The comparative study for the free energies and the weak hydrogen bond interactions in the enantio-determining transition state structures indicates that the cycloaddition reaction of 4a with 2a tends to produce 3a rather than the isomer with the help of more weak hydrogen-bond interactions, as depicted in Fig. S4 (Supporting information). In contrast, in Fig. S2, based on the structure of Int1A to complete the following ring formation reaction, the energy barrier for this process of Int1A-TS1A-2A'-Int2A' is significantly 27.6 kcal/mol higher than that shown in Fig. 1. As with [cat-5]+ recovery, the final product 3a is formed at the end of this reaction. The most distinctive feature of this type of reaction is the presence of hydrogen bonds in each intermediate and each transition state in Fig. 1, and detailed information about these intermolecular hydrogen bonds is shown in Fig. S3 (Supporting information). From the perspective of NBO charges and energy barriers, the intermediate Int1A, formed by 4a, [2a]- and [cat-5]+ in Fig. 1 and Fig. S2, tends to acquire an H+ from the reaction environment before the ring formation in this type of enantioselective cycloaddition reaction of 4a with 2a catalyzed by cat-5. This observation also applies to the second case, the cycloaddition reactions catalyzed by cat-6, as shown in Figs. S5 and S6 (Supporting information). In Fig. S5, by comparing the energy barriers between pathway B and pathway B', the same reactants 4a and 2a in Fig. 1 and Fig. S2 undergo the enantioselective cycloaddition reaction catalyzed by [cat-6]+ via an energy barrier of 18.1 kcal/mol to generate the final product 3a'.
In conclusion, an enantioselective phosphonium-salt catalyzed formal [5 + 3] cycloaddition reaction between alkenyl azomethine imines and γ-butenolides was developed under mild conditions, which efficiently accessed to diverse coronal polyheterocyclic compounds with high yields and ee values. DFT calculations elucidated the detailed mechanisms of the enantioselective cycloaddition reaction catalyzed by dipeptide-phosphonium salts. The hydrogen bonds between reactants and catalysts play an important role in enantio-controlling the absolute configurations of the products. In-vitro biological activity testing experiments also revealed the potential applications of these coronal polyheterocyclic compounds in drug discovery. All these results exhibited that alkenyl azomethine imines could be applied as powerful building blocks to generate interesting structures as useful compounds in drug research, which will pave a new avenue for the applications of alkenyl azomethine imines. In addition, enantioselective [3 + 2] cycloaddition reactions were also investigated catalyzed by dipeptide-phosphonium salts to generate chiral polyheterocyclic compounds.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jun Liu: Investigation. Zhaoyu Feng: Investigation. Renming Pan: Methodology. Xiaolong Yu: Resources. Meijuan Zhou: Formal analysis. Gang Zhao: Conceptualization. Hongyu Wang: Writing – review & editing, Writing – original draft, Supervision, Project administration, Funding acquisition, Conceptualization.
Financial support from National Natural Science Foundation of China (Nos. 21871282, 22377113, 22301309), Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0590000), National Key Research and Development Program of China (No. 2023YFA0914502), Taishan Scholar Program of Shandong Province (No. tsqn202306103, tsqn202306026), Postdoctoral Fellowship Program of CPSF (No. GZC20232509), Distinguished Young Scholars of Shandong Province (Overseas) (No. 2022HWYQ-004), Shandong Postdoctoral Science Foundation (No. SDBX2023044), Qingdao Postdoctoral Science Foundation (No. QDBSH20230202048), and the Fundamental Research Funds for the Central Universities (Ocean University of China) were gratefully acknowledged. The work was carried out at the National Supercomputer Center in Tianjin, and the calculations were performed on Tianhe's new-generation supercomputer.
Supplementary material associated with this article can be found, in the online version, at doi:
L.M. Lima, B.N.M.d. Silva, G. Barbosa, E.J. Barreiro, Eur. J. Med. Chem. 208 (2020) 112829.
M. Mora-Ochomogo, C.T. Lohans, RSC Med. Chem. 12 (2021) 1623–1639. doi: 10.1039/d1md00200g
M. Typers, G.D. Wright, Nat. Rev. Microbiol. 17 (2019) 141–155.
V.P. Sandanayaka, A.S. Prashad, Curr. Med. Chem. 9 (2002) 1145–1165. doi: 10.2174/0929867023370031
R.J. Worthington, C. Melander, J. Org. Chem. 78 (2013) 4207–4213. doi: 10.1021/jo400236f
N. Brown, E. Jacoby, Mini-Rev. Med. Chem. 6 (2006) 1217–1229. doi: 10.2174/138955706778742768
S. Shaabani, C.G. Neochoritis, A. Twarda-Clapa, et al., Med. Chem. Commun. 8 (2017) 1046–1052.
Y. Hu, D. Stumpfe, J. Bajorath, J. Med. Chem. 60 (2017) 1238–1246. doi: 10.1021/acs.jmedchem.6b01437
L.O. Davis, Org. Prep. Proced. Int. 45 (2013) 437–464. doi: 10.1080/00304948.2013.834769
M. Garg, M. Chauhan, P.K. Singh, J.M. Alex, R. Kumar, Eur. J. Med. Chem. 97 (2015) 444–461.
U. Grošelj, J. Svete, ARKIVOC 6 (2015) 175–205. doi: 10.3998/ark.5550190.p009.129
T. Hashimoto, K. Maruoka, Chem. Rev. 115 (2015) 5366–5412. doi: 10.1021/cr5007182
C. Nájera, J. Sansano, M. Yus, Org. Biomol. Chem. 13 (2015) 8596–8636.
L. Wei, X. Chang, C.J. Wang, Acc. Chem. Res. 53 (2020) 1084–1100. doi: 10.1021/acs.accounts.0c00113
R. Shintani, Y.T. Soh, T. Hayashi, Org. Lett. 12 (2010) 4106–4109. doi: 10.1021/ol101700v
R. Huber, R. Bigler, A. Mezzetti, Organometallics 34 (2015) 3374–3384. doi: 10.1021/acs.organomet.5b00357
H.Y. Wang, C.W. Zheng, Z. Chai, J.X. Zhang, G. Zhao, Nat. Commun. 7 (2016) 12720.
R. Shintani, G.C. Fu, J. Am. Chem. Soc. 125 (2003) 10778–10779.
W. Chen, W. Du, Y.Z. Duan, et al., Angew. Chem. Int. Ed. 46 (2007) 7667–7670. doi: 10.1002/anie.200702618
M. Hori, A. Sakakura, K. Ishihara, J. Am. Chem. Soc. 136 (2014) 13198–13201. doi: 10.1021/ja508441t
Q. Du, J.M. Neudorfl, H.G. Schmalz, Chem. Eur. J. 24 (2018) 2379–2383. doi: 10.1002/chem.201800042
G.L. Chai, E.Z. Yao, R.H. Liu, J. Chang, Org. Lett. 24 (2022) 6449–6454. doi: 10.1021/acs.orglett.2c02597
B. Gu, S. Wu, H. Xu, et al., Chin. Chem. Lett. 32 (2021) 672–675.
N. Luo, Z. Zheng, Z. Yu, Org. Lett. 13 (2011) 3384–3387. doi: 10.1021/ol201139w
M. Zhang, F. Wu, H. Wang, J. Wu, W. Chen, Adv. Synth. Catal. 359 (2017) 2768–2772. doi: 10.1002/adsc.201700387
T. Imaizumi, Y. Yamashita, S. Kobayashi, J. Am. Chem. Soc. 134 (2012) 20049–20052. doi: 10.1021/ja311150n
L.P. Kong, N.K. Li, S.Y. Zhang, et al., Org. Biomol. Chem. 12 (2014) 8656–8670.
R.H. Li, G.L. Chai, X. Wang, H.Y. Deng, J. Chang, J. Org. Chem. 88 (2023) 16566–16580. doi: 10.3390/ijms242316566
X. Wu, Q. Liu, Y. Liu, et al., Adv. Synth. Catal. 355 (2013) 2701–2706. doi: 10.1002/adsc.201300383
D. Cao, J. Zhang, H. Wang, G. Zhao, Chem. Eur. J. 21 (2015) 9998–10002. doi: 10.1002/chem.201501806
H. Hong, H. Wang, C. Zheng, G. Zhao, Chin. Chem. Lett. 32 (2021) 708–712.
F. Weinhold, J. Comb. Chem. 33 (2012) 2363–2379. doi: 10.1002/jcc.23060
V. Rai, I.N.N. Namboothiri, Eur. J. Org. Chem. 2006 (2006) 4693–4703. doi: 10.1002/ejoc.200600505
C. Nájera, J. Sansano, M. Yus, Org. Biomol. Chem. 13 (2015) 8596–8636.
B.M. Trost, E. Gnanamani, J.S. Tracy, C.A. Kalnmals, J. Am. Chem. Soc. 139 (2017) 18198–18201. doi: 10.1021/jacs.7b11361
T. Sakai, S. i. Hirashima, Y. Matsushima, et al., Org. Lett. 21 (2019) 2606–2609. doi: 10.1021/acs.orglett.9b00574
B.M. Trost, E. Gnanamani, C.A. Kalnmals, C.I.J. Hung, J.S. Tracy, J. Am. Chem. Soc. 141 (2019) 1489–1493. doi: 10.1021/jacs.8b13367
A.R. Choudhury, S. Mukherjee, Chem. Soc. Rev. 49 (2020) 6755–6788.
H. Wang, C. Zheng, G. Zhao, Chin. J. Chem. 37 (2019) 1111–1119. doi: 10.1002/cjoc.201900276
H. Li, H. Liu, H. Guo, Adv. Synth. Catal. 363 (2021) 2023–2036. doi: 10.1002/adsc.202001604
S. Fang, Z. Liu, T. Wang, Angew. Chem. Int. Ed. 62 (2023) e202307258.

Scheme 1 Applications of N,N-bicyclic pyrazolidine-3-ones and new reactivities of N,N′-cyclic azomethine imines for generating chiral polyheterocyclic compounds. TBAB: tetrabutylammonium bromide.

Scheme 2 The substrate scope of the cycloaddition. Condition A: the reactions were carried out with 1 (0.10 mmol), 2 (0.15 mmol), cat-5 (5 mol%), and K3HPO4 (0.20 mmol) in anisole (1.0 mL) at room temperature. Condition B: the reactions were carried out with 1 (0.10 mmol), 2 (0.15 mmol), cat-6 (5 mol%) and K3HPO4 (0.20 mmol) in anisole (1.0 mL) at room temperature. a 5-Methylfuran-2(3H)-one was used. The ee values were determined by HPLC analysis. The dr was determined by 1H NMR analysis of the crude products. Yields of 3 were isolated yields.

Scheme 3 The substrate scope of the [3 + 2] cycloaddition reactions. The reactions were carried out with 4 (0.10 mmol), 2 (0.15 mmol), cat-7 (5 mol%), and K2CO3 (0.2 mmol) in toluene (1.0 mL) at room temperature. a 5-Methylfuran-2(3H)-one was used. b 4-Methylfuran-2(5H)-one was used. The ee values were determined by HPLC analysis. The dr was determined by 1H NMR analysis of the crude products. Yields of 5 were isolated yields.

Scheme 5 In-vitro biological activity testing. Bioactive investigations were screened by CCK-8 assay (see Supporting information for detailed conditions).

Figure 1 The energy profile for pathway A of the enantioselective cycloaddition reaction catalyzed by cat-5. The relative free energies in anisole as solvent are given in kcal/mol. The partial hydrogen atoms are hidden for clarity.
Table 1. Optimization of the reaction conditions.a
|
||||||
| Entry | Cat. | Base | Solvent | 3a/3′ | Yield (%)b | ee of 3a (%)c |
| 1d | cat-1 | K2CO3 | Toluene | 4/6 | 64 | 50 |
| 2d | cat-1 | K2CO3 | TBME | 6/4 | 57 | 48 |
| 3 | cat-1 | K2CO3 | CH2Cl2 | 8/2 | 64 | 38 |
| 4 | cat-1 | K2CO3 | Anisole | 6/4 | 75 | 70 |
| 5 | cat-1 | K2HPO4 | Anisole | > 20/1 | 93 | 80 |
| 6 | cat-1 | K3PO4 | Anisole | 3/7 | 55 | 71 |
| 7 | cat-1 | Cs2CO3 | Anisole | 2/8 | 46 | 72 |
| 8 | cat-2 | K2HPO4 | Anisole | > 20/1 | 95 | 70 |
| 9 | cat-3 | K2HPO4 | Anisole | > 20/1 | 79 | 79 |
| 10 | cat-4 | K2HPO4 | Anisole | > 20/1 | 60 | 7 |
| 11 | cat-5 | K2HPO4 | Anisole | > 20/1 | 94 | 93 |
| 12 | cat-6 | K2HPO4 | Anisole | > 20/1 | 95 | -87 |
| 13e | cat-6 | K2HPO4 | Anisole | > 20/1 | 93 | -87 |
| a Unless otherwise specified, the reaction was carried out with 1a (0.10 mmol), 2a (0.15 mmol), catalyst (5 mol%), and base (0.2 mmol) in anisole (1.0 mL) at room temperature for 48 h. b Isolated yields of product. c Determined by chiral HPLC analysis. d The reaction time is 7 days. e With 3.0 mol% cat-6. dr of 3a were > 20:1. |
||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
