
Figure 1.
(a) Full-optimized structure of the 2D Ni-TABQ monolayer. (b) Band structure of 2D Ni-TABQ monolayer. (c) Electron localization function (ELF) diagram of 2D Ni-TABQ monolayer.
Two-dimensional conjugated coordination polymer monolayer as anode material for lithium-ion batteries: A DFT study
Xin-Tong Zhao , Jin-Zhi Guo , Wen-Liang Li , Jing-Ping Zhang , Xing-Long Wu

The global increasing demand and severe depletion of fossil fuels along with attendant environmental pollution and human's health concerns have resulted in the desire for green, clean and abundant energy sources such as wind energy, solar energy and tidal energy. However, such green energies are unstable and noncontinuous, which promotes the rapid development of a series of energy storage systems. Among a variety of energy storage systems, rechargeable lithium-ion batteries (LIBs) as a kind of high-efficiency electrochemical energy storage device, have been widely put into use in portable electronic equipment [1–4] and large-scale smart grids [5,6] on account of their wide voltage window, high energy density, and excellent electrochemical performance [7–9].
Anode is one of the most important components of lithium-ion batteries and plays a crucial role in lithium-ion batteries. However, commercial graphite anode, whose capacity is only 372 mAh/g [10], is not only difficult to meet people's growing requirements and expectations for energy storage systems, but also to satisfy the increasing market demand for rapid charge and discharge process due to its disadvantageous diffusion barrier. Therefore, the performance of conventional lithium-ion batteries based on graphite anode is not prominent enough. Hence, the pursue for a new anode material with good conductivity, large capacity and excellent rate performance has become a research hotspot.
Organic electrode materials have caught increasing attention owing to their unique advantages such as adjustable molecular structure, ascendant specific capacity and economic effectiveness [11–15]. For the past few years, conjugated coordination polymers (CCPs) consisting of π-d conjugated organic ligands and metal ions have been proved to be a class of active electrode materials for energy storage systems. Li's group synthesized an organic layered anode Ni-TABQ by π-d hybridization, which provides a high capacity of about 469.5 mAh/g at 100 mA/g and 345.4 mAh/g at 8 A/g. The large capacity can be maintained for 100 cycles, with almost 100% coulomb efficiencies [16]. Meantime, Li's team reported hexagonal MOF nanowire arrays, NiRu-HTP. The utilization of NiRu-HTP in the Li–O2 battery contributed to significantly reduced charge/discharge polarization of 0.76 V and exceptional cyclability [17]. They also reported two-dimensional Fe-TABQ as electrode materials for lithium-ion batteries, which delivered a remarkable specific capacity of 251.1 mAh/g and maintained capacity retentions over 95% after undergoing 200 cycles at diverse current densities [18]. Using TABQ as organic ligand and M2+ (M = Co, Ni, and Cu) as metal ligands, Wang's group reported a series of one-dimensional organic electrode materials in which 1D Ni-TABQ showed higher conductivity and the best electrochemical performance. 1D Ni-TABQ can be used as cathode and anode material to construct high-performance all-organic symmetric LIB with energy density up to 142 Wh/kg and stable cyclability of up to 100 times at 200 mA/g [19]. Besides, Wang's group also has been successful in synthesizing a variety of CCPs, which have been successfully applied to ionic battery systems [20–22]. Cheng's group reported two isostructural two-dimensional CPs, namely [M2(pztc)(H2O)6]n (M = Co or Ni), delivered excellent reversible capacities and stable cycling performance as anodes in lithium-ion batteries [23]. Cheng's group also reported that linking the mononuclear complex [Ni(PBIM)2(HIPA)] to a one-dimensional coordination polymer [Ni(PBIM)(HIPA)]n via coordination bonds through a facile bottom-up assembly approach is capable of markedly enhancing the lithium storage capacity from 554 mAh/g to 1025 mAh/g at 100 mA/g [24]. Lately, they synthesized three two-dimensional Sn-organic materials with enhanced lithium storage performance by manipulation of π-aromatic conjugation of the ligands [25]. Fan's group synthesized multilayer COF by a facile solvothermal route. The COF delivered exceptional potassium storage performance, including the excellent reversible capacity of 229.5 mAh/g at 0.2 A/g and superior cycling stability of 106.1 mAh/g at 5.0 A/g after 2000 cycles [26].
In terms of the dimensions of materials, two-dimensional materials have developed with a surprising momentum in recent years. Since the emergence of graphene in 2004 [27], all kinds of two-dimensional (2D) materials [28–32] with atomic thickness have received widely attention in virtue of their distinctive properties, such as unprecedented physicochemical properties, large specific surface area, preeminent mechanical properties, abundant active sites, short ion diffusion paths and good electrical conductivity. By means of experiments as well as theoretical research, a variety of two-dimensional materials have been proved to have excellent performance in multifarious applications, including energy storage systems [33–40].
Combining the advantages of organic electrode materials and two-dimensional materials, we propose a two-dimensional conjugated coordination polymer 2D Ni-TABQ monolayer as a promising candidate anode material for lithium-ion batteries. 2D Ni-TABQ monolayer not only maintains the enhanced conductivity of bulk conjugated coordination polymer with a band gap of only 0.2 eV, but also possesses the advantageous characteristics of two-dimensional materials, such as rapid ion migration. Furthermore, 2D Ni-TABQ monolayer has a maximum lithium storage capacity of 722 mAh/g, nearly twice that of commercial graphite anode, suitable open-circuit voltage range (0.22–1.12 V) and superior stability. More interestingly, we have confirmed the feasibility of 2D Ni-TABQ monolayer exfoliation from the bulk multilayer Ni-TABQ by calculating the atom binding energy and exfoliation energy.
Our study was based on first-principles density functional theory (DFT) and was performed by Vienna ab initio simulation package (VASP) [41]. The projected augmented wave (PAW) approach was chosen for description of the ion–electron interaction [42]. The generalized gradient approximation (GGA) in the Perdew-Burke-Ernzerhof (PBE) functional was selected to describe the exchange and correlation functional [43,44]. A kinetic-energy cutoff of 450 eV was employed for the plane-wave basis set. A 5 × 5 × 1 grid for k-point sampling was used for geometry optimization and other calculations. Both the lattice constants and atom positions were fully optimized until the residual forces were less than 0.02 eV/Å. Furthermore, the convergence criterion for energy was set to 10−5 eV. For the 2D Ni-TABQ monolayer, a vacuum space of 30 Å along z direction has been set in order to avoid interaction between layers. Besides, DFT-D2 method was considered in all calculations to give an account of the van der Waals interaction [45]. We applied the climbing image nudged elastic band method (CI-NEB) [46,47] to determine the diffusion barriers and minimum energy pathways of Li diffusion on the 2D Ni-TABQ surface. In all of the CI-NEB calculations, nine images were created. Bader charge code was applied to quantify the amount of charge transfer between Li atoms and 2D Ni-TABQ monolayer.
The atom binding energy (Eb) is defined as Eq. 1 [48]:
|
|
(1) |
where EC, EH, EN, EO, ENi, and ENi-TABQ are the total energies of the C atom, H atom, N atom, O atom, Ni atom and 2D Ni-TABQ monolayer, respectively.
The exfoliation energy per unit area Ee is given by means of the undermentioned equation [49]:
|
|
(2) |
where Eiso(n) refers to the energy of an isolated n-layer Ni-TABQ slab in vacuum (here, n = 1), Ebulk represents the energy of bulk Ni-TABQ composed of m layers, thus Ebulk/m corresponds to the energy of the bulk Ni-TABQ per layer, and A is the in-plane area of the bulk unit cell.
The average adsorption energy of the Li atoms on the 2D Ni-TABQ monolayer is defined as Eq. 3:
|
|
(3) |
where ELix1/2Ni-TABQ, ENi-TABQ, and ELi represent the total energies of the 2D Ni-TABQ monolayer adsorbed by x Li atoms, the 2D Ni-TABQ monolayer, and the single Li atom in its corresponding bulk structure, respectively.
The theoretical specific capacity of 2D Ni-TABQ monolayer is calculated by the undermentioned equation:
|
|
(4) |
where n indicates the number of valence electrons of the Li atom (n = 1), xmax represents the maximum concentration of Li atoms adsorbed of 2D Ni-TABQ monolayer, F is the Faraday constant (26, 801 mAh/mol), and MNi-TABQ is the molecular weight of 2D Ni-TABQ monolayer.
The open-circuit voltage (OCV) of the process can be estimated by dint of the equation given below:
|
|
(5) |
where ELix1/2Ni-TABQ refers to the energy of Lix1/2Ni-TABQ, x indicates the number of Li atoms adsorbed of 2D Ni-TABQ monolayer, n represents the valence state of Li (n = 1) and ELi represents the energy of the single Li atom in its corresponding bulk structure, respectively.
The charge density difference of single Li atom adsorbed at the most favorable site on the 2D Ni-TABQ monolayer by dint of the undermentioned equation:
|
|
(6) |
where ρNi−TABQ and ρLi@Ni−TABQ stand for the electron charge densities of the 2D Ni-TABQ monolayer before and after the adsorption of single Li atom, and ρLi refers the electron charge density of single Li atom isolated in the system by keeping the unaltered condition and lattice parameters.
The phonon dispersion calculation was carried out using a 1 × 2 × 1 supercell by the PHONOPY code. The elastic constants were calculated to verify the mechanical stability. The Visualization for Electronic and STructure Analysis (VESTA) package was applied for visualization of structures as well as charge density difference [50].
First of all, Fig. 1a shows the top view of 2D Ni-TABQ monolayer after full optimization. Obviously, 2D Ni-TABQ monolayer is a kind of two-dimensional material with atomic thickness (Fig. S1 in Supporting information). Then, we explored the possibility to exfoliate an atomic monolayer from Ni-TABQ in its corresponding bulk phase. The exfoliation energy is one of the most powerful tools to evaluate its exfoliation feasibility. The exfoliation energy is defined as the amount of energy required to strip away the top layer from the surface of the bulk structure, serving as an indicator for the level of difficulty of exfoliating a monolayer from corresponding bulk structure. The exfoliation energy of bulk Ni-TABQ is 4.7 meV/Å2, which indicates that bulk Ni-TABQ is facile to peel off. Moreover, the atom binding energy of 2D Ni-TABQ monolayer is 5.5 eV/atom, which is higher than MX3 (1.92–4.36 eV/atom, M = Sc, As, Y, Sb, Bi; X = Cl, Br, I) [48] and MoS2 (5.0–5.1 eV/atom) [51,52], which indicates stronger binding strength and weaker interlayer interaction of Ni-TABQ. In a word, the exfoliation energy and the atom binding energy collectively illustrate the feasibility of forming 2D Ni-TABQ monolayer and suggest that 2D Ni-TABQ monolayer can be obtained by means of laboratory facilities, just like two-dimensional materials that can be stripped experimentally, such as graphene, MoS2 and WS2 [53–55].
As everyone knows, the electronic property of anode materials plays a vital role in their application in rechargeable battery systems. So then, the electronic property of 2D Ni-TABQ monolayer was calculated and discussed. Fig. 1b reveals the band structure of 2D Ni-TABQ monolayer, and it is found that 2D Ni-TABQ monolayer is a semiconductor with a direct band gap of about 0.2 eV. Such a small band gap can suggest that the electronic property of 2D Ni-TABQ monolayer would take place an evident shift from semiconductor property to metallic property after Li adsorption. The electron localization function (ELF) diagram is an efficient tool to analyze the bonding characteristics between atoms. The ELF value of larger (> 0.5) corresponds to the covalent bond or core electrons, while the ionic bond is represented by the ELF value of smaller (< 0.5), and the ELF value of 0.5 represents the existence of the metallic bond. As shown in Fig. 1c, the coexistence of covalent bonds and metallic bonds on 2D Ni-TABQ monolayer is apparent that demonstrates the stability of 2D Ni-TABQ monolayer. The structural stability is a prerequisite for material application in batteries. Mechanical stability of materials is still important as well as electronic property when applied as anode materials. In the field of theoretical calculation, elastic constants are always powerful tools to verify the mechanical stability of materials. We further calculated the in-plane planar elastic constants, Cij, of 2D Ni-TABQ monolayer. As can be observed from Table 1, the elastic constants of 2D Ni-TABQ monolayer satisfy the Born-Huang criteria (C11C22 > C122, C66 > 0), which prove its excellent mechanical stability. It can also be seen from the elastic constants that 2D Ni-TABQ monolayer possesses anisotropic property (C11 ≠ C22). Furthermore, we calculated the phonon spectrum of 2D Ni-TABQ monolayer to evaluate its dynamic stability. As shown in Fig. S2 (Supporting information), there is no imaginary phonon mode, which justifies that 2D Ni-TABQ monolayer is dynamically stable.
The appropriate adsorption energy of adatoms on the substrate is an important parameter to evaluate the properties of anode materials for LIBs. Hence, after investigating the properties of 2D Ni-TABQ monolayer itself, the interaction between Li atoms and substrate was further explored to evaluate the potential of 2D Ni-TABQ monolayer as anode material for lithium-ion batteries. In order to conduct the following study, the most favorable adsorption site of single Li atom on 2D Ni-TABQ monolayer was primarily detected. According to the symmetry of 2D Ni-TABQ monolayer, a total of three kinds of adsorption sites have been examined, namely, the top sites of atom (M sites), the top sites of the bond center (B sites) and the hollow sites (H sites). However, after full optimization, it is turned out that all the M sites and B sites are unstable, so only four unequal adsorption sites remained. As shown in Fig. 2a, these four sites are named H1 (the top site at the center of a six-membered ring between two polymer chains), H2 (the top site at the center of a ten-membered ring between two polymer chains), H3 (the top site at the center of a five-membered ring of a single polymer chain), and H4 (the top site at the center of C6 ring). The adsorption energy (Ea) of single Li atom on 2D Ni-TABQ monolayer is calculated by Eq. 3. According to the definition of adsorption energy, the more negative the adsorption energy is, the more favorable the adsorption of Li atom on the substrate is. The specific adsorption energies of each adsorption sites are shown in Fig. 2b. The adsorption energies are −1.17 eV (H1), −1.13 eV (H2), −0.47 eV (H3), and 0.09 eV (H4), respectively. The adsorption energy of Li atom adsorbed at the H1 site has the lowest energy, indicating that the H1 site is the most stable adsorption site.
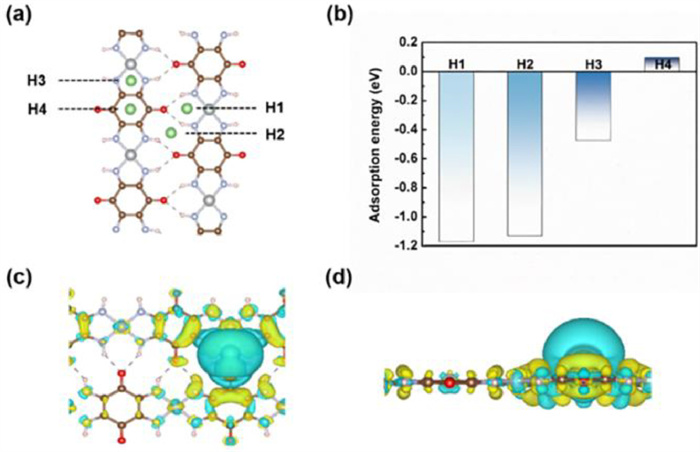
The development of high-efficiency batteries is highly dependent on the capabilities of anode materials, especially during the charge and discharge process, which is largely driven by the dynamic characteristic of electron. Furthermore, the mechanism behind the adsorption behavior of single Li atom on 2D Ni-TABQ monolayer was further investigated by calculating the charge density difference. Figs. 2c and d have shown the charge density difference of 2D Ni-TABQ monolayer after the adsorption of single Li atom. As shown in these figures, yellow shape represents the electron accumulation and bluish shape represents electron depletion. There is a significant electron depletion around the Li atom and electron accumulation around substrate, indicating obvious electron transfer between Li atom and 2D Ni-TABQ monolayer. The charge transfer between Li atom and substrate has been quantified by means of Eq. 6, and the Bader charge analysis [56] reveals that 0.90 e is transferred from Li atom to the substrate, Li contributes almost all valence electron and exists in the form of cation.
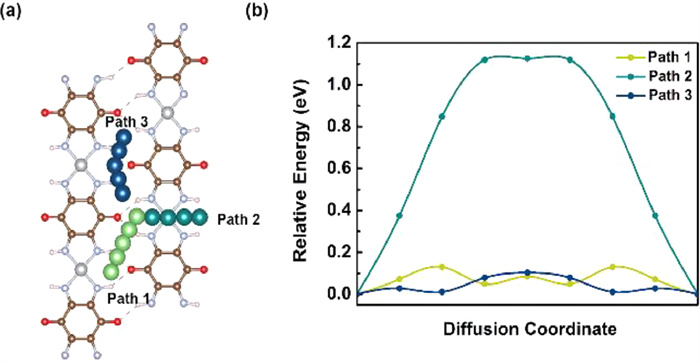
The rate and cyclic properties are very vital for lithium-ion batteries which are strongly associated with the diffusion performance of Li ions. Therefore, the diffusion behavior of Li ions on the surface of 2D Ni-TABQ monolayer was further explored by using CI-NEB method. According to the symmetry of 2D Ni-TABQ monolayer and the most stable adsorption site (H1), two diffusion paths were initially of consideration, named path 1 and path 2, respectively.
Path 1 indicates the diffusion of lithium ions between neighboring H1 sites between polymer chains, and path 2 describes the diffusion of lithium ions across chain between neighboring H1 sites. Figs. 3a and b show the top diagram of these two diffusion paths and their corresponding relative energy profiles. It can be seen that the interchain diffusion process path 1 is the more favorable diffusion path, with a diffusion barrier of merely 0.13 eV. Since the adsorption energy at H2 site is extremely close to that at H1 site, interchain diffusion between adjacent H2 sites was also taken into account, namely path 3. Path 3 has a diffusion barrier of 0.10 eV, even smaller than path 1. Such small diffusion barriers of path 1 and path 3 prove the rapid ion diffusion rate that is beneficial to the rate performance and cyclic performance, suggesting that 2D Ni-TABQ monolayer be capable of being potential candidate for anode materials of Li-ion batteries. Table 2 lists the comparison of the diffusion barriers between 2D Ni-TABQ monolayer and other previously reported two-dimensional materials [57–64]. It can be seen that the diffusion barriers of 2D Ni-TABQ monolayer are smaller than that of other two-dimensional materials, indicating its excellent diffusion performance.
 DownLoad:
CSV
DownLoad:
CSV

|
The specific capacity of anode materials is one of the vital indexes to evaluate the properties of anode materials. In a rocking chair battery, Li ions are constantly transferred between the cathode and anode during charge and discharge process. Li ions are gradually inserted into both sides of 2D Ni-TABQ monolayer to highlight the insertion process (Fig. 4). Firstly, Li ions occupy all the most favorable adsorption sites on one side of 2D Ni-TABQ monolayer, which corresponds to the stoichiometric ratio of Li2Ni-TABQ. On this basis, Li ions occupy H1 sites on the other side of 2D Ni-TABQ monolayer, labeled as Li4Ni-TABQ. This structure of Li4Ni-TABQ is used as the initial structure to explore the most favorable adsorption site for the next layer of Li adsorption. The H3 site becomes the most favorable adsorption site instead of H1 site. 2D Ni-TABQ monolayer can successfully adsorb the second layer of Li on one side, labeled as Li6Ni-TABQ. However, when the second layer of Li is adsorbed on the other side, the adsorption energy becomes positive, indicating that 2D Ni-TABQ monolayer cannot achieve complete two-layer Li adsorption. The stoichiometric ratio corresponding to the maximum adsorption concentration is Li6Ni-TABQ, which corresponds to the maximum theoretical capacity of 722 mAh/g calculated by Eq. 4. According to the structure of Li6Ni-TABQ, the average distance between the first Li layer and the substrate, namely the adsorption height, is 1.21 Å, while the adsorption height between the second Li layer and the substrate is 1.54 Å. Detailed structural information such as lattice parameters, bond length, is listed in Table S1 (Supporting information).

Next, the open-circuit voltage of 2D Ni-TABQ monolayer as anode material for Li-ion batteries was studied. Figs. 5a and b show the adsorption energies and open-circuit voltage at different Li adsorption concentrations, respectively. Apropos of the adsorption energies, from Li0.25Ni-TABQ to the maximum adsorption concentration configuration Li6Ni-TABQ, the adsorption energies are always negative, indicating that Li atoms are more inclined to form Li ions rather than Li metal clusters. As for the open-circuit voltage, for different lithium adsorption concentrations, the open-circuit voltage ranges from 1.12 V to 0.22 V, and the average open-circuit voltage is 0.57 V, which meets the requirement of the anode material for the open-circuit voltage.

Finally, the electron behavior after Li atoms adsorbed on 2D Ni-TABQ monolayer was further studied. Fig. S3 (Supporting information) shows the band structure of Li6Ni-TABQ. It can be seen that during the lithium insertion process, there is a clear transition from semiconductor property to metallic property. Table 3 shows the charge transfer of the adsorption by single Li atom and different adsorption concentrations of Li. It can be seen that Li atoms contribute almost all valence electron and exist in the form of ions.
DownLoad:
CSV

|
In summary, we have discussed the prospective applicability of 2D Ni-TABQ monolayer as anode material of lithium-ion batteries on the basis of density functional theory method. First of all, the elastic constants of 2D Ni-TABQ monolayer satisfy the Born-Huang criteria, which indicates its excellent mechanical stability. There is no imaginary mode in phonon dispersion curve, which justifies that 2D Ni-TABQ monolayer is dynamically stable. Then, we have discussed the feasibility of exfoliation from bulk multilayer Ni-TABQ to form 2D Ni-TABQ monolayer in the light of exfoliation energy and the atom binding energy. Additionally, the electronic property of 2D Ni-TABQ monolayer is acceptable because of its pretty small band gap (0.2 eV). Most important of all, lithium storage on both sides of 2D Ni-TABQ monolayer can achieve theoretical specific capacity of 722 mAh/g, which nearly twice that of commercial graphite anode in terms of lithium-ion batteries. Apart from the storage property, the diffusion property of Li-ion on the surface of 2D Ni-TABQ monolayer is likewise preeminent due to its exceeding favorable diffusion barriers of 0.10 eV and 0.13 eV which significantly smaller relative to many other 2D anode materials. The open-circuit voltage of 2D Ni-TABQ monolayer is demonstrated within the appropriate range between from 1.12 V to 0.22 V. Based on our results, 2D Ni-TABQ monolayer is an excellent candidate for anode material of lithium-ion batteries.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work is financially supported by the National Natural Science Foundation of China (No. 52173246), Natural Science Foundation of Jilin Province (No. 20220508141RC), Double-Thousand Talents Plan of Jiangxi Province (No. jxsq2023102005), 111 Project (No. B13013), Education Department of Jilin Province (No. JJKH20221154KJ), and Shccig-Qinling Program.
Supplementary material associated with this article can be found, in the online version, at doi:
J.W. Choi, D. Aurbach, Nat. Rev. Mater. 1 (2016) 16013. doi: 10.1038/natrevmats.2016.13
C.F. Liu, Z.G. Neale, G.Z. Cao, Mater. Today 19 (2016) 109–123. doi: 10.1016/j.mattod.2015.10.009
S. Hy, H.D. Liu, M.H. Zhang, et al., Energy Environ. Sci. 9 (2016) 1931–1954. doi: 10.1039/C5EE03573B
F. Su, X.C. Hou, J.Q. Qin, Z.S. Wu, Batter. Supercaps 3 (2020) 10–29. doi: 10.1002/batt.201900153
T.Y. Liu, K.C. Kim, R. Kavian, S.S. Jang, S.W. Lee, Chem. Mater. 27 (2015) 3291–3298. doi: 10.1021/acs.chemmater.5b00314
X.G. Zhong, Y.Q. Wei, Y.P. Guo, et al., Electrochim. Acta 336 (2020) 135729. doi: 10.1016/j.electacta.2020.135729
D.C. Lin, Y.Y. Liu, Y. Cui, Nat. Nanotech. 12 (2017) 194–206. doi: 10.1038/nnano.2017.16
H.Y. Cha, J.H. Kim, H.Y. Lee, et al., Adv. Mater. 32 (2020) 2003040. doi: 10.1002/adma.202003040
L.Q. Zhang, C.X. Zhu, S.C. Yu, D.H. Ge, H.S. Zhou, J. Energy Chem. 66 (2022) 260–294. doi: 10.1016/j.jechem.2021.08.001
Y. Wen, K. He, Y.J. Zhu, et al., Nat. Commun. 5 (2014) 4033. doi: 10.1038/ncomms5033
C. Vaalma, D. Buchholz, M. Weil, S. Passerini, Nat. Rev. Mater. 3 (2018) 18013. doi: 10.1038/natrevmats.2018.13
Y. Lu, J. Chen, Nat. Rev. Chem. 4 (2020) 127–142. doi: 10.1038/s41570-020-0160-9
S. Lee, G. Kwon, K. Ku, et al., Adv. Mater. 30 (2018) 1870312. doi: 10.1002/adma.201870312
P. Poizot, J. Gaubicher, S. Renault, et al., Chem. Rev. 120 (2020) 6490–6557. doi: 10.1021/acs.chemrev.9b00482
C. Fang, Z. Ye, Y.J. Wang, et al., J. Mater. Chem. A 7 (2019) 22398–22404. doi: 10.1039/c9ta07403a
L.B. Wang, Y.X. Ni, X.S. Hou, et al., Angew. Chem. Int. Ed. 59 (2020) 22126–22131. doi: 10.1002/anie.202008726
Q.L. Lv, Z. Zhu, Y.X. Ni, et al., J. Am. Chem. Soc. 144 (2022) 23239–23246. doi: 10.1021/jacs.2c11676
J.R. Geng, Y.X. Ni, Z. Zhu, et al., J. Am. Chem. Soc. 145 (2023) 1564–1571. doi: 10.1021/jacs.2c08273
K. Li, J. Yu, Z.J. Si, et al., Chem. Eng. J. 450 (2022) 138052. doi: 10.1016/j.cej.2022.138052
K. Fan, C. Fu, Y. Chen, et al., Adv. Sci. 10 (2023) 2205760. doi: 10.1002/advs.202205760
Y.C. Wu, Y. Zhang, Y. Chen, et al., Energy Environ. Sci. 14 (2021) 6514–6525. doi: 10.1039/d1ee02414k
Y. Chen, Q. Zhu, K. Fan, et al., Angew. Chem. Int. Ed. 60 (2021) 18769–18776. doi: 10.1002/anie.202106055
J.W. Liu, L. Zhang, H.H. Li, et al., Sci. China Chem. 62 (2019) 602–608. doi: 10.1007/s11426-018-9370-0
J. Du, Y. Li, H.W. Liu, et al., ACS Appl. Mater. Interfaces 11 (2019) 25863–25869. doi: 10.1021/acsami.9b06114
J.W. Liu, J.L. Jiang, Q.Y. Zhou, et al., eScience 3 (2023) 100094. doi: 10.1016/j.esci.2023.100094
Z.H. Su, J.H. Huang, R.H. Wang, et al., J. Colloid Interf. Sci. 639 (2023) 7–13. doi: 10.1016/j.jcis.2023.02.012
K.S. Novoselov, A.K. Geim, S.V. Morozov, et al., Science 306 (2004) 666–669. doi: 10.1126/science.1102896
P. Vogt, P.D. Padova, C. Quaresima, et al., Phys. Rev. Lett. 108 (2012) 155501. doi: 10.1103/PhysRevLett.108.155501
W.L. Lu, H.Y. Nan, J.H. Hong, et al., Nano Res. 7 (2014) 853–859. doi: 10.1007/s12274-014-0446-7
W.B. Li, L.J. Kong, C.Y. Chen, et al., Sci. Bull. 63 (2018) 282–286. doi: 10.1016/j.scib.2018.02.006
A.J. Mannix, X.F. Zhou, B. Kiraly, et al., Science 350 (2015) 1513–1516. doi: 10.1126/science.aad1080
K.S. Novoselov, D. Jiang, F. Schedin, et al., Natl. Acad. Sci. U.S.A. 102 (2005) 10451–10453. doi: 10.1073/pnas.0502848102
H.R. Jiang, W. Shyy, M. Liu, et al., J. Mater. Chem. A 5 (2017) 672–679. doi: 10.1039/C6TA09264K
S.F. Lei, X.F. Chen, B.B. Xiao, W.T. Zhang, J. Liu, ACS Appl. Mater. Interfaces 11 (2019) 28830–28840. doi: 10.1021/acsami.9b07219
H.W. Tao, M. Zhou, R.X. Wang, et al., Adv. Sci. 5 (2018) 1801021. doi: 10.1002/advs.201801021
T.T. Yu, P.F. Gao, Y. Zhang, S.L. Zhang, Appl. Surf. Sci. 486 (2019) 281–286. doi: 10.1016/j.apsusc.2019.05.019
G. Barik, S. Pal, J. Phys. Chem. C 125 (2021) 8980–8992. doi: 10.1021/acs.jpcc.1c02238
N. Khossossi, D. Singh, A. Banerjee, et al., ACS Appl. Energy Mater. 4 (2021) 7900–7910. doi: 10.1021/acsaem.1c01247
X. Guo, C.D. Wang, W.J. Wang, et al., Nano Res. Energy 1 (2022) e9120026. doi: 10.26599/nre.2022.9120026
X.L. Liu, Z.T. Liu, W. Yang, et al., Mater. Today Chem. 26 (2022) 101002. doi: 10.1016/j.mtchem.2022.101002
G. Kresse, J. Furthmüller, Phys. Rev. B 54 (1996) 11169. doi: 10.1103/PhysRevB.54.11169
P.E. Blöchl, Phys. Rev. B 50 (1994) 17953. doi: 10.1103/PhysRevB.50.17953
G. Kresse, J. Furthmüller, Comp. Mater. Sci. 6 (1996) 15–50. doi: 10.1016/0927-0256(96)00008-0
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865. doi: 10.1103/PhysRevLett.77.3865
S. Grimme, J. Comput. Chem. 25 (2004) 1463–1473. doi: 10.1002/jcc.20078
G. Henkelman, B.P. Uberuaga, H. Jónsson, J. Chem. Phys. 113 (2000) 9901. doi: 10.1063/1.1329672
G. Henkelman, H. JónssonJ, Chem. Phys. 113 (2000) 9978.
P. Liu, F. Lu, M.K. Wu, et al., J. Mater. Chem. C 5 (2017) 9066–9071. doi: 10.1039/C7TC03003G
J.H. Jung, C.H. Park, J. Ihm, Nano Lett. 18 (2018) 2759–2765. doi: 10.1021/acs.nanolett.7b04201
K. Momma, F. Izumi, J. Appl. Cryst. 41 (2008) 653–658. doi: 10.1107/S0021889808012016
G.X. Wang, R. Pandey, S.P. Karna, WIREs Comput. Mol. Sci. 7 (2017) e1280. doi: 10.1002/wcms.1280
S. Ahmad, S. Mukherjee, Graphene 3 (2014) 52–59. doi: 10.4236/graphene.2014.34008
M.Z. Cai, D. Thorpe, D.H. Adamson, H.C. Schniepp, J. Mater. Chem. 22 (2012) 24992–25002. doi: 10.1039/c2jm34517j
H.C. Lin, J.W. Wang, Q.Q. Luo, et al., J. Alloy Compd. 699 (2017) 222–229. doi: 10.1016/j.jallcom.2016.12.388
J.P. Liu, H.B. Liu, W.C. Peng, et al., Chem. Eng. J. 431 (2022) 133286.
W. Tang, E. Sanville, G. Henkelman, J. Phys. Condens. Matter 21 (2009) 084204. doi: 10.1088/0953-8984/21/8/084204
Y. Wu, Z. Li, J.H. Hou, Appl. Surf. Sci. 599 (2022) 153958. doi: 10.1016/j.apsusc.2022.153958
V. Shukla, R.B. Araujo, N.K. Jena, R. Ahuja, Nano Energy 41 (2017) 251–260. doi: 10.1016/j.nanoen.2017.09.026
X.M. Zhang, Z.M. Yu, S.S. Wang, et al., J. Mater. Chem. A 4 (2016) 15224–15231. doi: 10.1039/C6TA07065E
W. Liu, X.T. Zu, Y.G. Zhou, H.X. Deng, Appl. Surf. Sci. 581 (2022) 152368. doi: 10.1016/j.apsusc.2021.152368
C.M. Zhang, Y.L. Jiao, T.W. He, et al., Phys. Chem. Chem. Phys. 19 (2017) 25886–25890. doi: 10.1039/C7CP04758D
Y. Wu, S.L. Wang, Y.Q. Xie, X. Ye, S.T. Sun, Appl. Surf. Sci. 574 (2022) 151296.
D.W. Zhou, Z. Wang, J.B. Cheng, C.Y. Pu, Processes 10 (2022) 1809. doi: 10.3390/pr10091809
K. Persson, V.A. Sethuraman, L.J. Hardwick, et al., J. Phys. Chem. Lett. 1 (2010) 1176–1180. doi: 10.1021/jz100188d

Figure 1 (a) Full-optimized structure of the 2D Ni-TABQ monolayer. (b) Band structure of 2D Ni-TABQ monolayer. (c) Electron localization function (ELF) diagram of 2D Ni-TABQ monolayer.

Figure 2 (a) The unequal sites in 2D Ni-TABQ monolayer. (b) Adsorption energies of single Li atom adsorbed at H1 site, H2 site, H3 site and H4 site on 2D Ni-TABQ monolayer. (c) Top and (d) side view of charge density difference of Li-adsorbed 2D Ni-TABQ monolayer at H1 site.

Figure 3 (a) Possible diffusion pathways for Li ions on 2D Ni-TABQ monolayer. (b) Calculated diffusion energies profiles of Li ions on 2D Ni-TABQ monolayer along different diffusion pathways. The light green, dark green and dark blue ball-chains are migration pathways of path 1, path 2 and path 3, respectively.

Figure 5 (a) The average adsorption energies and (b) the OCV profile of LixNi-TABQ under certain Li concentrations.
Table 2. Comparison of diffusion barriers of 2D Ni-TABQ monolayer as anode for LIBs with available literature precedents.
|
|
 下载: 导出CSV
下载: 导出CSV
Table 3. The charge transferred of the adsorption by single Li atom and different adsorption concentrations of Li.
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

