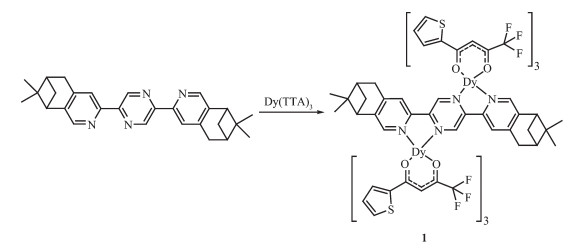
Scheme 1.
双核镝配合物1的合成路线
Scheme 1.
Synthesis route of complex 1

分子磁性材料以其在信息存储与处理方面的独特价值与应用前景已经引起了全世界科学家的关注[1-6]。区别于传统磁体以单原子或离子作为构筑基元,分子磁体是以分子为基本结构单元,通过氢键、π-π键等非共价键作用、静电作用、范德华力等弱相互作用和(或)配位键构筑而成。分子磁体通常具有以下优点:(1)合成方法相对简单、多样;(2)易获得单晶,适于开展磁性机制的理论研究;(3)体积小,相对密度轻,结构多样化,易于裁剪、调控和加工成型。如何将原子组合成各式各样的分子,进而合理组装以形成理想结构,并获得优良性质,无疑是化学家们长期面临的机遇和挑战[7]。
稀土离子因其独特电子结构受到化学及材料等领域科学家的广泛关注[8-10]。近年来,在配合物分子中引入稀土离子(如:Tb3+,Dy3+,Ho3+,Er3+,Gd3+等)来制备高各向异性能垒单离子磁体受到广泛关注,并取得了很大的进展[11-16]。然而,现有单离子磁体的阻塞温度还远低于室温,离实际应用还有很大的距离。对于稀土单离子磁体,通常其具有较高的磁各向异性能垒,但在低温下量子隧穿效应非常明显,所以如何有效降低或阻止量子隧穿效应,是制备高阻塞温度稀土单离子磁体面临的最大挑战。为此,在合成多核簇合物和提高各向异性能垒上深入研究,与其它学科进一步融合,运用新的测试手段和提出新理论将会促进单离子磁体研究的发展。本文报道了采用蒎烯联吡啶基吡嗪配体与Dy(Ⅲ)-二酮反应制备的一例新的双核镝配合物[Dy2(PY)(TTA)3] (1),并通过X射线单晶衍射确定了其单晶结构,表征了配合物的磁性。
药品均为市售分析纯,未经预处理直接使用。蒎烯吡啶基吡嗪配体(PY)和Dy(TTA)3·2H2O的合成参考文献报道的方法[17-18],所使用的手性源为Aldrich公司生产的(1R)-(-)-桃金娘烯醛。磁性测试使用单晶培养实验获得的晶体样品,采用美国MPMS-SQUIDXL-7超导量子干涉仪,直流磁化率的测量温度为1.8~300 K,测量磁场为2 000 Oe,磁化测量温度为1.8~10.0 K,测量磁场为1.0~7.0 T。交流磁化率的测量频率为1、10、100、498、997、1 488 Hz,振荡场为3.0 Oe,直流场为0 Oe。
将Dy(TTA)3·2H2O(0.861 g,1 mmol)、蒎烯吡啶基吡嗪配体(PY)(0.221 g,0.5 mmol)、乙醇(30 mL)置于单口瓶中,常温搅拌反应12h,析出白色固体,过滤。所得配合物粉末溶解在二氯甲烷和乙醇的混合物溶剂中(体积比为1:1),常温下缓慢挥发4周后得到无色晶体1(0.375 g,收率:36%)。元素分析按C76H54Dy2N4F18O12S6计算值(%):C,44.00;H,2.62;N,2.70。实测值(%):C,44.12;H,2.69;N,2.76。
单晶结构分析采用德国Bruker SMART APEX-Ⅱ CCD X射线单晶衍射仪,Mo Kα(λ=0.071 073 nm),管电流50 kV,管电流30 mA,室温下采用SMART程序收集衍射数据。运用SAINT程序还原数据和SADABS程序进行经验吸收校正[19-20]。结构解析和精修采用SHELXTL程序[21]。
CCDC:1874301。
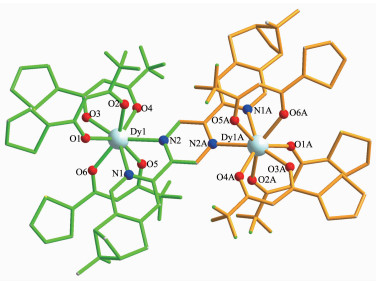
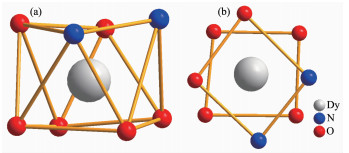
双核镝配合物1的晶体结构如图 1所示,晶体参数列于表 1,键长键角列于表 2。配合物1是中性双核Dy(Ⅲ)配合物,结晶于空间群P21/n,每个分子是由1个蒎烯吡啶基吡嗪配体、6个β-二酮阴离子、2个Dy(Ⅲ)阳离子组成。配合物在结晶过程中,手性中心蒎烯部分发生了内消旋化,导致配合物结晶在一个有心的空间群。双核镝配合物1的不对称单元结构(图 1中绿色化学键部分)包括1个Dy(TTA)3·0.5PY单元,Dy1、N1、N2、O1、O2、O3、O4、O5和O6通过对称操作分别得到Dy1A、N1A、N2A、O1A、O2A、O3A、O4A、O5A和O6A(Symmetry codes:A:1-x,1-y,2-z)。Dy(Ⅲ)离子与每个β-二酮阴离子的2个氧原子进行配位,另外2个位置被手性配体中2个氮原子占据,从而形成了Dy(Ⅲ)离子的八配位构型。每个配合物分子中的2个Dy(Ⅲ)离子通过共轭体系吡嗪环桥联,位于吡嗪环的两侧。Dy-O键键长从0.228 2(6)到0.234 5(7) nm不等。Dy-N键键长分别是0.252 5(7)到0.256 9(8) nm。由于键长各不相等,使Dy(Ⅲ)离子处于扭曲的反四棱柱配位构型中。如图 2所示,O1-O2-N3-N4(底面)和O3-O4-O5-O6(顶面)分别构成了反四棱柱的2个基本面。通常,标准的反四棱柱的2个基本面是相互平行的,一个面相对另一个面而言,旋转了45°,而在1的结构中,旋转了42.1°。文献中报道的Dy(Ⅲ)离子构型有3种形式:双帽三棱柱,扭曲反四棱柱,扭曲十二面体,配合物1中的Dy(Ⅲ)离子的配位构型非常接近反四棱柱,属于扭曲反四棱柱构型[18]。
H atoms are omitted for clarity; Symmetry codes: A: 1-x, 1-y, 2-z
 下载:
导出CSV
下载:
导出CSV
| Empirical formula | C76H54Dy2N4F18O12S6 |
| Formula weight | 2 074.59 |
| Temperature / K | 296(2) |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a / nm | 1.544 30(14) |
| b / nm | 1.542 76(13) |
| c / nm | 1.823 90(16) |
| β/(°) | 104.595(2) |
| Volume / nm3 | 4.205 2(6) |
| Z | 2 |
| Dc / (g · cm-3) | 1.638 |
| F(000) | 2 048 |
| θ range / (°) | 1.548~24.999 |
| Reflection collected | 23 494 |
| Goodness-of-fit on F2 | 0.917 |
| Unique reflection | 7 401 |
| Rint | 0.112 7 |
| Reflection with I>2σ(I) | 3 882 |
| Number of parameter | 534 |
| R1a, wR2b [I>2σ(I)] | 0.062 4, 0.137 0 |
| R1, wR2 (all data) | 0.141 4, 0.159 9 |
| (Δρ)max, (Δρ)min / (e·nm-3) | 1 027, -676 |
| a R1=Σ(|Fo|-|Fc|)/Σ|Fo|; b wR2=[Σw(Fo2-Fc2)2/Σw(Fo2)2]1/2 | |
下载:
导出CSV
| Dy1-O1 | 0.234 5(7) | Dy1-O2 | 0.232 3(7) | Dy1-O4 | 0.233 6(7) |
| Dy1-O3 | 0.228 2(6) | Dy1-O6 | 0.230 4(6) | Dy1-N2 | 0.256 9(8) |
| Dy1-O5 | 0.233 0(7) | Dy1-N1 | 0.252 5(7) | ||
| O5-Dy1-O4 | 79.5(2) | O5-Dy1-O1 | 145.1(2) | O1-Dy1-O6 | 78.1(2) |
| O4-Dy1-O1 | 133.4(2) | O5-Dy1-O6 | 71.9(2) | O4-Dy1-O6 | 124.2(2) |
| O5-Dy1-O2 | 139.0(2) | O4-Dy1-O2 | 74.9(3) | O3-Dy1-O2 | 88.7(2) |
| O1-Dy1-O2 | 71.8(2) | O6-Dy1-O2 | 149.0(2) | N1-Dy1-N2 | 63.2(2) |
| O5-Dy1-O3 | 113.8(3) | O4-Dy1-O3 | 73.2(2) | O4-Dy1-N1 | 142.3(2) |
| O1-Dy1-O3 | 74.5(2) | O6-Dy1-O3 | 76.2(2) | O6-Dy1-N1 | 78.3(2) |
| O5-Dy1-N1 | 80.4(3) | O1-Dy1-N1 | 76.4(3) | O3-Dy1-N1 | 144.5(2) |
| O2-Dy1-N1 | 1O1.1(3) | O5-Dy1-N2 | 72.8(2) | O4-Dy1-N2 | 80.4(2) |
| O1-Dy1-N2 | 117.4(2) | O6-Dy1-N2 | 131.0(2) | O2-Dy1-N2 | 71.7(2) |
(a) Slightly distorted square antiprism; (b) Pseudo-D4d local symmetry
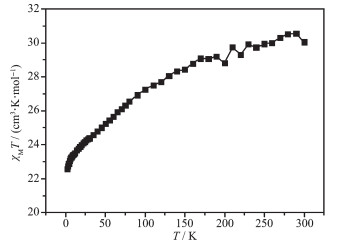
在100 Oe的外加磁场下,表征了配合物1粉末样品在1.8~300 K温度范围的变温磁化率曲线(图 3)。结果表明,配合物1的χMT数值在室温时为30.58 emu·K·mol-1(大于2个顺磁Dy离子的理论值28.34 emu·K·mol-1,J=15/2,g=4/3),并随着温度的不断降低而逐渐下降,在1.8 K达到最低值22.49 emu·K·mol-1。配合物1的χMT数值随着温度的下降应归因于Dy离子大的轨道贡献导致斯塔克能级的去热布居过程。
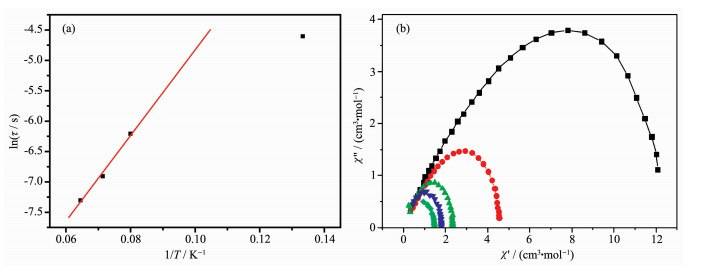
在零场下,测试了配合物1粉末样品的低温交流磁化率曲线(图 4)。测试结果表明,配合物1交流磁化率的实部和虚部在0~20 K的温度下都显示出明显的频率依赖现象,表明材料为单分子磁体。如图 4b所示,配合物的交流磁化率虚部在100~1 488 Hz的频率下,7.5~15.5 K的温度下都出现了频率依赖峰值。配合物的交流磁化率虚部在频率(1~10 Hz)及温度(小于6 K)较低时,曲线随温度下降而上升,表明在这些条件下存在快速弛豫过程。通常这些快速弛豫过程来自于材料的量子隧穿效应,并且可以通过外加磁场或者掺杂抗磁性离子(如Gd离子)的样品来抑制[22]。因为低频下(100 Hz)交流磁化率的频率依赖峰值与高温下的峰值明显偏离,所以我们将配合物交流磁化率虚部(498~1 488 Hz)高温部分的频率依赖峰值应用阿伦尼乌斯公式(τ=τ0exp[Ueff/(kBT)])曲线拟合,可以得到单分子磁体高温部分的能垒为71.1 K,指前因子τ0为6.59 μs(图 5a)。材料的单离子磁体慢磁弛豫行为同时也被样品在外加直流磁场为0 Oe时的Cole-Cole曲线所证实。如图 5b所示,将交流磁化率在1.8、5、10、13、16 K及1~1 488 Hz频率下测定的χ′对χ″值作图,即可得到单分子磁体的Cole-Cole曲线。曲线通过德拜公式拟合得到参数a值分别为0.31,0.30,0.15,0.15,0.12(a值接近于0为单弛豫过程),表明此单分子磁体在低温下可能存在多个弛豫过程。该单分子磁体的弛豫中高温部分应该属于Orbach过程,低温部分应包含有Raman过程。
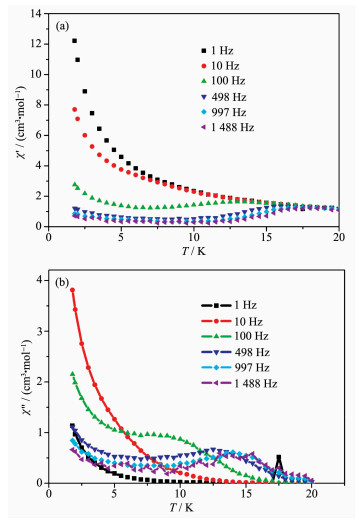
(a) In-phase; (b) Out-of-phase
我们通过将含手性蒎烯吡啶基的嘧啶类衍生物与Dy(TTA)3反应,并采用溶液法生长晶体,制备了双核镝配合物1。配合物的单晶结构解析表明,配合物1中一个蒎烯基团的构型发生了手性翻转。磁性测试表明配合物1在无外加直流磁场下表现出明显的慢磁弛豫性质。通过阿伦尼乌斯公式曲线拟合可以得到单分子磁体的能垒为71.1 K,指前因子τ0为6.59 μs。上述结果为基于Dy(Ⅲ)配合物的稀土功能材料的设计合成提供了有益的新思路。
游效曾.分子材料光电功能化合物. Shanghai:Shanghai Science and Technology Press, 2001.YOU Xiao-Zeng. Molecular-Based Materials:Opto-electronic Functional Compounds. Shanghai:Shanghai Science and Technology Press, 2001.
Yamanouchi M, Chiba D, Matsukura F, et al. Nature, 2004, 428(6982):539-542 doi: 10.1038/nature02441
Saitoh E, Miyajima H, Yamaoka T, et al. Nature, 2004, 432(7034):203-206 https://www.ncbi.nlm.nih.gov/pubmed/15538364
Yang L, Li J, Pu T C, et al. Dalton Trans., 2017, 46(20):6670-6676 doi: 10.1039/C7DT00753A
Wu D Q, Shao D, Wei X Q, et al. J. Am. Chem. Soc., 2017, 139(34):11714-11717 doi: 10.1021/jacs.7b07008
Bogani L, Wernsdorfer W. Nat. Mater., 2008, 7(3):179-186 doi: 10.1038/nmat2133
Leuenberger M N, Loss D. Nature, 2001, 410(6830):789-793 doi: 10.1038/35071024
Hussain B, Savard D, Burchell T J, et al. Chem. Commun., 2009(9):1100-1102 doi: 10.1039/b818295g
Liu X Q, Zhang Y, Shi W, et al. Inorg. Chem., 2018, 57(21):13409-13414 doi: 10.1021/acs.inorgchem.8b01981
王庭玮, 陈洪进, 张蕤, 等.无机化学学报, 2018, 34(6):1007-1017 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180601WANG Ting-Wei, CHEN Hong-Jin, ZHANG Rui, et al. Chinese J. Inorg. Chem., 2018, 34(6):1007-1017 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180601
Ishikawa N, Sugita M, Ishikawa T, et al. J. Am. Chem. Soc., 2003, 125(29):8694-8695 doi: 10.1021/ja029629n
Ge J Y, Wang H Y, Li J, et al. Dalton Trans., 2017, 46(10):3353-3362 doi: 10.1039/C7DT00298J
Jiang S D, Wang B W, Su G, et al. Angew. Chem. Int., Ed., 2010, 49(41):7448-7451 doi: 10.1002/anie.v49:41
Tang J, Hewitt I, Madhu N T, et al. Angew. Chem. Int. Ed., 2006, 45(11):1729-1733 doi: 10.1002/(ISSN)1521-3773
Takamatsu S, Ishikawa T, Koshihara S Y, et al. Inorg. Chem., 2007, 46(18):7250-7252 doi: 10.1021/ic700954t
Li D P, Wang T W, Li C H, et al. Chem. Commun., 2010, 46(17):2929-2931 doi: 10.1039/b924547b
Li X L, Gao Y L, Feng X L, et al. Dalton Trans., 2012, 41(38):11829-11835 doi: 10.1039/c2dt31007d
Li X L, Chen C L, GaoY L. Chem. Eur. J., 2012, 18:14632-14637 doi: 10.1002/chem.201201190
SAINT-Plus, Vers. 6.02, Bruker Analytical X-ray System, Madison, WI, 1999.
Sheldrick G M. SADABS, an Empirical Absorption Correction Program, Bruker Analytical X-ray Systems, Madison, WI, 1996.
Sheldrick G M. SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997.
Li D P, Zhang X P, Wang T W, et al. Chem. Commun., 2011, 47(24):6867-6869 doi: 10.1039/c1cc11659b

图 1 双核镝配合物1的晶体结构图
Figure 1 Crystal structure of complex 1
H atoms are omitted for clarity; Symmetry codes: A: 1-x, 1-y, 2-z

图 2 双核镝配合物1中Dy(Ⅲ)离子的配位构型
Figure 2 Coordination environment of Dy(Ⅲ) ion
(a) Slightly distorted square antiprism; (b) Pseudo-D4d local symmetry

图 3 双核镝配合物1的变温磁化率曲线
Figure 3 Variable-temperature magnetic susceptibility of complex 1

图 4 双核镝配合物1在3 Oe交流磁场和0 Oe直流磁场下的交流磁化率信号
Figure 4 Dynamic magnetic susceptibilities of complex 1 measured in a 3 Oe AC magnetic field with a zero Oe DC-field
(a) In-phase; (b) Out-of-phase

图 5 (a) 双核镝配合物1的阿伦尼乌斯拟合曲线; (b)在3 Oe交流磁场和0 Oe直流磁场下的Cole-Cole图
Figure 5 (a) Arrhenius fitting curve for complex 1; (b) Cole-Cole plots for ac susceptibility of complex 1 collected under a 3 Oe AC magnetic field with a zero Oe DC-field
表 1 双核镝配合物1的晶体学数据
Table 1. Crystallographic data of complex 1
| Empirical formula | C76H54Dy2N4F18O12S6 |
| Formula weight | 2 074.59 |
| Temperature / K | 296(2) |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a / nm | 1.544 30(14) |
| b / nm | 1.542 76(13) |
| c / nm | 1.823 90(16) |
| β/(°) | 104.595(2) |
| Volume / nm3 | 4.205 2(6) |
| Z | 2 |
| Dc / (g · cm-3) | 1.638 |
| F(000) | 2 048 |
| θ range / (°) | 1.548~24.999 |
| Reflection collected | 23 494 |
| Goodness-of-fit on F2 | 0.917 |
| Unique reflection | 7 401 |
| Rint | 0.112 7 |
| Reflection with I>2σ(I) | 3 882 |
| Number of parameter | 534 |
| R1a, wR2b [I>2σ(I)] | 0.062 4, 0.137 0 |
| R1, wR2 (all data) | 0.141 4, 0.159 9 |
| (Δρ)max, (Δρ)min / (e·nm-3) | 1 027, -676 |
| a R1=Σ(|Fo|-|Fc|)/Σ|Fo|; b wR2=[Σw(Fo2-Fc2)2/Σw(Fo2)2]1/2 | |
 下载: 导出CSV
下载: 导出CSV
表 2 双核镝配合物1的部分键长和键角
Table 2. Selected bond lengths (nm) and angles (°) for complex 1
| Dy1-O1 | 0.234 5(7) | Dy1-O2 | 0.232 3(7) | Dy1-O4 | 0.233 6(7) |
| Dy1-O3 | 0.228 2(6) | Dy1-O6 | 0.230 4(6) | Dy1-N2 | 0.256 9(8) |
| Dy1-O5 | 0.233 0(7) | Dy1-N1 | 0.252 5(7) | ||
| O5-Dy1-O4 | 79.5(2) | O5-Dy1-O1 | 145.1(2) | O1-Dy1-O6 | 78.1(2) |
| O4-Dy1-O1 | 133.4(2) | O5-Dy1-O6 | 71.9(2) | O4-Dy1-O6 | 124.2(2) |
| O5-Dy1-O2 | 139.0(2) | O4-Dy1-O2 | 74.9(3) | O3-Dy1-O2 | 88.7(2) |
| O1-Dy1-O2 | 71.8(2) | O6-Dy1-O2 | 149.0(2) | N1-Dy1-N2 | 63.2(2) |
| O5-Dy1-O3 | 113.8(3) | O4-Dy1-O3 | 73.2(2) | O4-Dy1-N1 | 142.3(2) |
| O1-Dy1-O3 | 74.5(2) | O6-Dy1-O3 | 76.2(2) | O6-Dy1-N1 | 78.3(2) |
| O5-Dy1-N1 | 80.4(3) | O1-Dy1-N1 | 76.4(3) | O3-Dy1-N1 | 144.5(2) |
| O2-Dy1-N1 | 1O1.1(3) | O5-Dy1-N2 | 72.8(2) | O4-Dy1-N2 | 80.4(2) |
| O1-Dy1-N2 | 117.4(2) | O6-Dy1-N2 | 131.0(2) | O2-Dy1-N2 | 71.7(2) |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
