Department of Physics and Synergetic Innovation Center for Quantum Effects and Applications, Key Laboratory of Low-Dimensional Quantum Structures and Quantum Control of Ministry of Education, Key Laboratory for Matter Microstructure and Function of Hunan Province, Hunan Normal University, Changsha 410081, China
b.
Hunan Joint International Laboratory of Advanced Materials and Technology for Clean Energy, Hunan University, Changsha 410082, China
* Corresponding authors at: Department of Physics and Synergetic Innovation Center for Quantum Effects and Applications
Key Laboratory of Low-Dimensional Quantum Structures and Quantum Control of Ministry of Education
Key Laboratory for Matter Microstructure and Function of Hunan Province
Received Date:
03 January 2022 Accepted Date:
17 February 2022 Revised Date:
23 January 2022 Available Online:
15 January 2023
Abstract:
Transition metal hydroxides/oxyhydroxides have recently emerged as highly active electrocatalysts for oxygen evolution reaction in alkaline water electrolysis, while have not yet been widely investigated for hydrogen evolution electrocatalysts owing to their unfavorable H*-adsorption, making it difficult to construct an overall-water-splitting cell for hydrogen production. In this work, we proposed a straightforward and effective approach to develop an efficient in-plane heterostructured CoOOH/Co(OH)2 catalyst via In-situ electrochemical dehydrogenation method, in which the dehydrogenated –CoOOH and Co(OH)2 at the surface synergistically boost the hydrogen evolution reaction (HER) kinetics in base as confirmed by high-resolution transmission electron microscope, synchrotron X-ray absorption spectroscopy, and electron energy loss spectroscopy. Due to the In-situ dehydrogenation of ultrathin Co(OH)2 nanosheets, the catalytic activity of the CoOOH/Co(OH)2 heterostructures is progressively improved, which exhibit outstanding hydrogen-evolving activity in base requiring a low overpotential of 132 mV to afford 10 mA/cm2 with very fast reaction kinetics after 60 h dehydrogenation. The gradually improved catalytic performance for the CoOOH/Co(OH)2 is probably due to the enhanced H*-adsorption induced by the synergistic effect of heterostructures and better conductivity of CoOOH relative to electrically insulating Co(OH)2. This work will open the opportunity for a new family of transition metal hydroxides/oxyhydroxides as active HER catalysts, and also highlight the importance of using in situ techniques to construct precious metal-free efficient catalysts for alkaline hydrogen evolution.
Electrocatalytic water splitting driven by renewable energies (e.g., solar, wind, geothermal energy) has widely regarded as a safe and environmentally friendly pathway for hydrogen production [1-6]. At present, water splitting is more favored in alkaline electrolyte by most people due to its compatibility with inexpensive and earth-abundant electrocatalysts and cheap electrolyzer construction [7-11]. However, most noble metal-free HER catalysts (e.g., transition metal sulfides [12-15], selenides [16-20], phosphides [21-26]) performing superior activity in acidic solution, possess unsatisfied HER performance in alkaline electrolytes owing to their poor capability for H2O adsorption/dissociation. Based on this point, intensive efforts have been devoted to further improving the catalytic performance by hybridizing these materials with transition metal hydroxides (e.g., Ni(OH)2, Co(OH)2, NiFe LDH) [27-29]. Here, transition metal hydroxides are reported to serve as OH− (generated by H2O electrolysis) adsorption sites rather than H*-adsorption centers, which indicates that the catalysts promoter of transition metal hydroxides alone exhibit inferior activity for HER due to the vital role of hydrogen binding in this process. In this sense, it is very promising if we can find out some useful routes to activate the reaction kinetics of transition metal hydroxides. Actually, it is interesting to note that transition metal oxyhydroxides as the intermediates in the transformation process from hydroxides to oxides have been demonstrated to be electrocatalytically active for HER [30, 31]. For example, Pillai et al. investigated the catalytic activity of CoOOH towards HER by calculating the adsorption energy of hydrogen atom, and the results suggest that CoOOH is a promising candidate for HER [31]. Therefore, as a missing piece in alkaline HER, heterostructures by integrating transition metal oxyhydroxides with hydroxides is highly expected to exhibit outstanding hydrogen-evolving activity considering better conductivity of CoOOH relative to Co(OH)2.
Recently, In-situ electrochemical activation has drawn considerable attention as a new pretreating strategy for tuning electrocatalytic activity owing to its simplicity, variable control and flexibility [32-36]. More importantly, the electrochemical activation strategy exerted on the as-obtained precursors would in situ create abundant active species at the surface, always keeping a strong binding force between the active species and conductive supports, which would further facilitate the electron transfer from the electrode to the catalyst surface and ensure structural stability during the durability tests [36, 37]. In addition, the local electronic structure and morphology of catalysts can be effectively modulated viain-situ electrochemical tuning technique [38, 39]. For example, Hu et al. reported an In-situ electrochemical activation method to pretreat Ni-based ligand 1, 4-benzenedithiol (Ni-BDT) nanosheets, which can be transformed into ultrathin metallic Ni (Ni0) nanosheets with trace sulfide (Sadsδ−) adsorbed on the surface under a cathodic potential [37]. Benefiting from the Ni0-Sadsδ− interface, the water dissociation process has been greatly promoted, thereby significantly enhancing HER activity in an alkaline electrolyte. Besides the In-situ electrochemical reduction, the In-situ electrochemical etching also can be utilized to regulate the activity of catalysts. For instance, Hu's group adopted a galvanostatical etching method to activate the OER performance of perovskite CoSn(OH)6 nanocubes via dissolving Sn hydroxides, creating O-vacancies and generating porous structures [38].
Inspired by the aforementioned In-situ strategies, we devoted to constructing porous cobalt-based in-plane oxyhydroxide/hydroxide (CoOOH/Co(OH)2) heterostructures with outstanding HER activity from electrochemically inert Co(OH)2via an In-situ electrochemical dehydrogenation/activation method. The electrochemically dehydrogenated hybrid catalyst shows a superior catalytic activity toward HER with a low overpotential of 132 mV to reach 10 mA/cm2, which is greatly decreased in comparison with that of pristine Co(OH)2 (464 mV). This unusual self-optimizing performance can be derived from the generation of CoOOH/Co(OH)2 heterostructures, in which Co(OH)2 acts as water adsorption/dissociation promoter and CoOOH serves as H*-adsorption site as well as the improved electrical conductivity and charge transfer of the as-obtained catalysts.
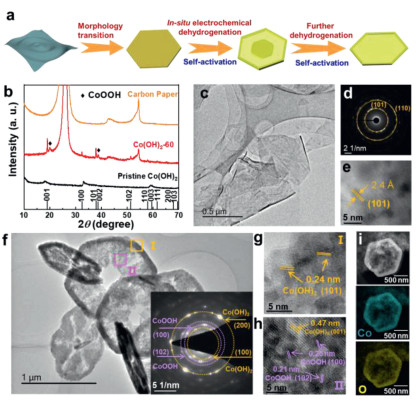
In order to fabricate an in-plane heterostructure between conductive CoOOH and Co(OH)2, we employed a novel strategy called In-situ electrochemical activation to dehydrogenate ultrathin Co(OH)2 nanosheets under a constant current density of −10 mA/cm2 in 1.0 mol/L KOH electrolyte. The activation process was performed using a three-electrode electrochemical cell Fig. 1a illustrates the evolution of morphology and component of Co(OH)2 nanosheets as efficient HER catalysts in the In-situ electrochemical dehydrogenation process Fig. 1b shows the X-Ray diffraction (XRD) patterns of pristine Co(OH)2 and Co(OH)2 after In-situ electrochemical dehydrogenation (under reduction current density of 10 mA/cm2) for 60 h (denoted as: Co(OH)2-60). Although tiny peaks shift induced by interlayer ions (NO3− and CO32−) can be observed, the diffraction peaks of pristine Co(OH)2 located at around 18.1°, 33.0°, and 59.4° can be indexed to (001), (100) and (003) planes of hexagonal Co(OH)2 (PDF #45–0031). After electrochemical dehydrogenation, the peaks also can be well maintained for Co(OH)2-1 and Co(OH)2-5 samples (Fig. S1 in Supporting information), while two new prominent diffraction peaks at around 19.8° and 38.7° can be observed for Co(OH)2-60, which indicates that CoOOH would be gradually generated with the prolonged activation time Fig. 1c gives the typical TEM image of pristine Co(OH)2, and it clearly shows a thin, smooth and flexible sheet-like morphology. The selected area electron diffraction (SAED) pattern shows well-defined diffractions rings, indicating the polycrystalline nature of Co(OH)2 (Fig. 1d). The two rings can be ascribed to (101) and (110) lattice planes of the hexagonal Co(OH)2 (space group P-3m1). As can be observed from high-resolution TEM (HRTEM) image (Fig. 1e), the lattice fringes are randomly oriented, and a lattice spacing of 0.24 nm can be attributed to (101) plane of Co(OH)2, which is in good agreement with the SAED result.
Figure 1
Figure 1.
(a) Schematic illustration of the In-situ electrochemical dehydrogenation/activation process. (b) A typical XRD pattern of pristine Co(OH)2, activated Co(OH)2, and carbon paper. (c) TEM image, (d) SAED pattern, (e) HRTEM image of pristine Co(OH)2. (f) TEM image, (g, h) HR-TEM images selected in areas I and II, (i) STEM elemental mapping of Co(OH)2–60.
The In-situ electrochemical dehydrogenation of Co(OH)2 nanosheets was conducted at an cathodic current density of 10 mA/cm2, and they possess different morphologies with the prolonged of time. At the early stage, the ultrathin sheets morphology gradually transforms into hexagonal nanodiscs (Fig. S2 in Supporting information). With the increase of time, the nanosheets disappear and the solid structures became core-shell nanodiscs with a rough surface (Fig. S3 in Supporting information). At last, the solid core began to disappear, showing a nanoplate or nanoring-like structure. Typically, Co(OH)2-60 presents a nanoplate morphology with some pores in the centers and thicker rings around the out border (Fig. 1f). The inset of Fig. 1f is the SAED pattern of Co(OH)2-60, in which the marked yellow rings can be ascribed to the (100) and (200) planes of Co(OH)2, while the red rings can be assigned to the (100) and (102) planes of CoOOH, corroborating the in situ formation of an in-plane heterostructure between CoOOH and Co(OH)2. To further verify the composition and crystal structure of the Co(OH)2-60, HRTEM images in the interior and exterior areas are selected and marked by yellow and purple squares (Fig. 1f, I and II). In the area I, the lattice fringes with a distance of 0.24 nm can be clearly observed, which is corresponding to (101) plane of Co(OH)2 (Fig. 1g). In area II, it not only shows the (001) plane of Co(OH)2, but also displays the (100) and (102) planes of CoOOH with d-spacings of 0.25 nm and 0.21 nm (Fig. 1h), which is consistent with the SAED and XRD patterns. In addition, the STEM elemental mappings reveal the uniform distribution of Co and O in the as-prepared Co(OH)2-60 (Fig. 1i). All of these above observations confirm the uniform formation of CoOOH during the dehydrogenation process.
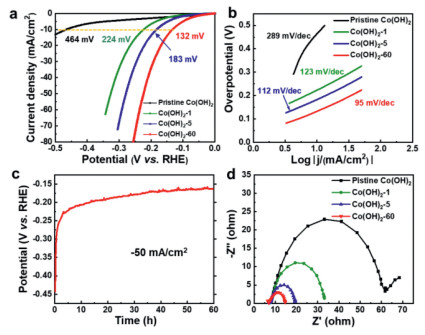
To verify the effects of heterostructures on the catalytic activity of CoOOH/Co(OH)2, electrochemical measurements of pristine Co(OH)2 and dehydrogenated Co(OH)2 after In-situ activation were conducted using a standard three-electrode system in 1.0 mol/L KOH. The pristine Co(OH)2 shows inferior HER performance with an overpotential of 464 mV at a current density of 10 mA/cm2, indicating that it is a truly poor candidate for HER catalysts (Fig. 2a). However, constant cathodic current density (10 mA/cm2) exerted on the as-prepared catalyst results in an activation and dehydrogenation process, which is evidenced by the dramatic decrease of overpotentials and gradual improvement of the catalytic activities. Specifically, the as-prepared Co(OH)2-1 and Co(OH)2-5 require overpotentials of 224 mV and 183 mV to reach a current density of 10 mA/cm2, respectively. In particular, Co(OH)2-60 displays very superior electrocatalytic activity with an overpotential of only 132 mV at 10 mA/cm2. A similar electrochemical performance improvement can be observed under continuous cyclic voltammetry between potentials of 0 and −0.3 V vs. RHE at a scan rate of 20 mV/s. After 1000 CV cycles, a low overpotential of only 256 mV is required for activated Co(OH)2, which is 210 mV lower than that of pristine Co(OH)2 (Fig. S4 in Supporting information). The CoOOH/Co(OH)2 heterostructures were In-situ generated during the process of continuous cyclic voltammetry measurement, as shown in (Fig. S5 Supporting information), in which two obvious diffraction peaks are detected belonging to CoOOH, suggesting the repeatable and credible dehydrogenation of Co(OH)2 to CoOOH at the surface for efficient HER catalysis. To exclude that the enhanced performance of the heterostructured catalyst is only originated from the increasement of CoOOH, electrochemical measurement has been conducted on pure CoOOH. As exhibited in (Fig. S6a Supporting information), the pure CoOOH requires overpotential of 453 mV to reach a current density of 10 mA/cm2, which is much higher than those of activated catalysts (Co(OH)2-1, Co(OH)2-5, and Co(OH)2-60). Moreover, the pure CoOOH shows sluggish catalytic kinetics as evidenced by the large Tafel slope of 159 mV/dec (Fig. S6b in Supporting information). This result reveals that pure CoOOH shows inferior HER performance and also demonstrates that the superior alkaline HER activity for the CoOOH/Co(OH)2 can be ascribed to the synergistic effect between the two components rather than the increase of CoOOH. The accelerated electrocatalytic kinetics of Co(OH)2 during the activation process is also evidenced by the distinct reduced Tafel slope, as exhibited in Fig. 2b. The Tafel slope of Co(OH)2-60 is 95 mV/dec, which is much lower than that of pristine Co(OH)2 (289 mV/dec). Although transition metal (oxy)hydroxides alone have been regarded as unqualified candidates for HER, our optimized Co(OH)2-based catalyst after In-situ electrochemical activation forming heterostructured CoOOH/Co(OH)2 exhibits superb catalytic activity, which outperforms most of the reported transition metal (oxy)hydroxides-based electrocatalysts tested under similar conditions (Table S1 in Supporting information), such as Ni(OH)2/MoS2 [40], Ni(OH)2/NiCo2O4 [41], CeO2/Co(OH)2 [42] and Co(OH)2@P-NiCo-LDH [43], and is even comparable to those of cobalt-based catalysts with much higher conductivity including phosphides (CoP@FeCoP and CoP@NC/graphene) [44, 45], selenides (p-CoSe2/CC and (Ni, Co)0.85Se) [46, 47], nitrides (NiCo2N/NF and Co3N) [48, 49], and sulfides (Co9S8 and CoSx/Ni3S2@NF) [50, 51]. The In-situ activation process can also be observed in the chronopotentiometry test conducted at a larger current density of −50 mA/cm2 (Fig. 2c), showing that the potential increases rapidly in the initial 5 h, and then increases gradually until the 60 h, which is consistent with the variation of the polarization curves in Fig. 2a. In addition, electrochemical impedance spectroscopy (EIS) was employed to explore the charge-transfer kinetics during the activation process. As presented in Fig. 2d, charge-transfer resistance of Co(OH)2 is continuously reduced from 56 Ω to 9 Ω with the increase of activation time, suggesting that In-situ electrochemical activation could efficiently improve the conductivity of the heterostructure, and accelerate the charge transfer kinetics between the electrolyte and catalysts.
Figure 2
Figure 2.
(a) iR-Corrected polarization curves in 1.0 mol/L KOH solution of various electrocatalysts as indicated (scan rate: 5 mV/s). (b) Tafel plots derived from the polarization curves in (a). (c) Chronopotentiometry curve of Co(OH)2 at a constant current density of −50 mA/cm2. (d) Nyquist plot measured at potential of −0.2 V vs. RHE.
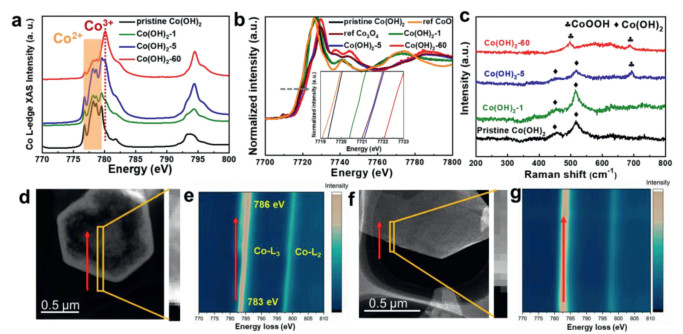
In order to gain an in-depth understanding of the mechanism behind this electrochemical dehydrogenation/activation process, we conducted a series of measurements including synchrotron X-ray absorption spectroscopy (XAS) and electron energy loss spectroscopy (EELS). The soft XAS can provide the useful information of electronic structure of Co in our as-prepared catalysts, as shown in Fig. 3a. Apparently, the L3 peaks located at 776.8, 778.1, 778.6, and 779.5 eV can be assigned to Co2+ in pristine Co(OH)2 [52]. While a peak located at 780.1 eV appears with the increase of activation time, which can be ascribed to Co3+ [52, 53]. It is clearly seen that the intensity of this peak increases with the prolonged activation time, suggesting the rising ratio of Co3+. In addition, X-ray absorption near-edge structure (XANES) spectra at the Co K-edge also demonstrate the existence of Co3+. As shown in Fig. 3b, the absorption edge is shifted to higher energy from the pristine Co(OH)2 to Co(OH)2-60, along with a broadening of the white line peak, revealing a distinct increase of the Co oxidation state. Although the XAS results suggest the presence of Co3+, it cannot probe the dynamic electronic structure of the catalyst during the electrocatalytic reaction. Therefore, In-situ Raman spectroscopy was conducted to directly monitor the structural transformation of Co(OH)2 under the electrochemical activation conditions. Fig. 3c shows Raman spectra of pristine Co(OH)2 and Co(OH)2 at selected activation time operated at cathodic current density of 10 mA/cm2 immersed in 1.0 mol/L KOH. Apparently, the bands located at 455 and 515 cm−1 can be assigned to the Co–O symmetric stretching mode (Ag) and O–Co–O bending mode derived from the Co(OH)2 nanosheets [54]. After 1 h activation, the bands of Co(OH)2 are retained and no band-shift or new bands can be observed. When the electrochemical activation time increases to 5 h, one new prominent band appears at around 688 cm−1 besides the band of Co(OH)2, which is ascribed to CoOOH [55, 56]. With prolonged activation time, the bands of Co(OH)2 gradually disappear. When the activation time increases to 60 h, bands of Co(OH)2 disappear and bands appearing at 497 cm−1 belongs to the CoOOH [54], indicating the In-situ formation of CoOOH under cathodic current operation, which is consistence with the ex-situ XAS result. To precisely investigate the distribution of CoOOH on the nanoplates, EELS mappings of pristine Co(OH)2 and Co(OH)2-60 were acquired from the edge to the interior of the nanoplates, as shown in Figs. 3d-g. Unlike the pristine Co(OH)2 that no obvious shift of L3 and L2 peaks can be detected, L3 and L2 peaks of Co(OH)2-60 gradually shift to higher energy losses from the border to the inner of the nanoplates, indicating that the percentage of the Co3+ (CoOOH) increases gradually from the exterior to the interior of the Co(OH)2-60 nanoplates, which is consistent with the HRTEM observations.
Figure 3
Figure 3.
(a) Co L-edge XAS and (b) Co K-edge XANES spectra of the pristine Co(OH)2 and Co(OH)2 after electrochemical activation. (c) In-situ Raman spectrum of Co(OH)2 under different activation stages. (d) Dark-field STEM image and (e) corresponding EELS mapping of Co(OH)2-60. (f) Dark-field STEM image and (g) corresponding EELS mapping of pristine Co(OH)2.
Based on the above characterizations, the evolution of morphology with electrochemical activation time for Co(OH)2 can be assigned to the combination of recrystallization and inside-out Ostwald ripening mechanism. Firstly, the pristine Co(OH)2 featuring ultrathin nanosheets morphology would gradually convert to nanoplates-like Co(OH)2via the recrystallization in an alkaline electrolyte [57]. As time goes on, the particles on the edges are easy to grow into large ones, because the recrystallization preferentially occurs at the solid-liquid interface, while the inner crystallites tend to dissolve owing to the loose packing [58]. Therefore, the core-shell like morphology would gradually form on the nanosheet owing to the growth of exterior part and consumption of the inner substances. With prolonged time, the solid core gradually evacuates and generates some pores in the center. As the HER reaction proceeding, the surface of Co(OH)2 would lose some H atoms and electrons and transform into CoOOH owing to the existence of dissolved oxygen in the electrolyte and strong concentration of OH− near the catalysts [59]. However, the edges of the nanoplates are difficult to lose electron under reductive current because the edges are always the active sites for HER. Therefore, the center of the nanoplates tend to lose electrons, thereby generating CoOOH/Co(OH)2 heterostructures. Consequently, the XAS and EELS results indicate that the as-prepared Co(OH)2 nanosheets experience distinct electronic structure reconstruction under realistic catalytic conditions forming heterostructured CoOOH/Co(OH)2, which is responsible for the In-situ electrochemical activation of Co(OH)2 for HER.
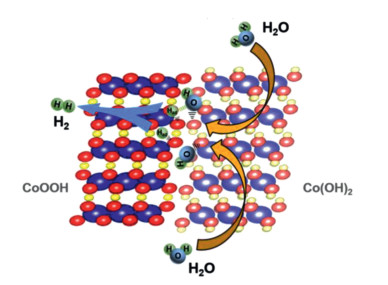
As has been reported, the adsorption/dissociation of H2O, which is regarded as a rate-limiting step in alkaline hydrogen evolution, prefers to proceed on the surfaces of transition metal hydroxides. Besides, CoOOH could serve as a more favorable site for hydrogen adsorption based on a small adsorption barrier of −0.45 eV [31]. Therefore, the superior alkaline HER activity for the CoOOH/Co(OH)2 is derived from the interface engineering. As shown in Fig. 4, the Co(OH)2 acts as a promoter for water adsorption and dissociation, while the produced hydrogen intermediates absorb on adjacent CoOOH and subsequently combine to give hydrogen gas. In this way, the CoOOH/Co(OH)2 heterostructures effectively reduce the energy barrier of the water dissociation and accelerates the alkaline HER process accordingly.
Figure 4
Figure 4.
Illustration of the HER mechanism on CoOOH/Co(OH)2 heterostructures in alkaline solution.
In this work, CoOOH/Co(OH)2 heterostructures have been designed and fabricated as an efficient alkaline HER electrocatalyst viain-situ electrochemical dehydrogenation strategy. The resultant CoOOH/Co(OH)2 heterostructures exhibit significantly enhanced HER performance with a small overpotential of 132 mV to afford 10 mA/cm2, which is 332 mV lower than that of pristine Co(OH)2. The excellent activity is probably attributed to the synergetic cooperation between Co(OH)2 and CoOOH, in which Co(OH)2 acts as H2O adsorption/dissociation promoter, and CoOOH serves as H* acceptor. We believe that our findings would provide novel insight into engineering heterostructured catalysts with outstanding HER performance in alkaline media.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This project is mainly funded by National Science Foundation of China (Nos. 12074116 and 52172197), the Youth 1000 Talent Program of China, Undergraduate Scientific Research Innovation Project of China (No. 202110542037), Science and Technology Innovation Platform (No. 2019RS1032), Major Program of Natural Science Foundation of Hunan Province of Hunan Province, and Hunan Normal University (Nos. 2021133, 21CSZ004 and 21CSZ029). H. Zhou also acknowledges the support from Hunan Joint International Laboratory of Advanced Materials and Technology for Clean Energy (No. 2020CB1007). Q. Zhou acknowledges the support from Science and Technology Innovation Program of Hunan Province (No. 2021RC2075).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.053.
[1]
Y. Zhang, Q. Zhou, J.X. Zhu, et al., Adv. Funct. Mater. 27 (2017) 1702317. doi: 10.1002/adfm.201702317
C.A. Triana, R. Moré, A.J. Bloomfield, et al., Matter 1 (2019) 1354–1369. doi: 10.1016/j.matt.2019.06.021
[57]
K. Ding, X. Zhang, J.P. Li, P. Yang, X. Cheng, CrystEngComm 19 (2017) 5780–5786. doi: 10.1039/C7CE01130J
[58]
F.Y. Kong, M. Li, X.Y. Yao, et al., CrystEngComm 14 (2012) 3858–3861. doi: 10.1039/c2ce25199j
[59]
K. Kongsawatvoragul, S. Kalasina, P. Kidkhunthod, M. Sawangphruk, Electrochim. Acta 324 (2019) 134854. doi: 10.1016/j.electacta.2019.134854
Figure 1
(a) Schematic illustration of the In-situ electrochemical dehydrogenation/activation process. (b) A typical XRD pattern of pristine Co(OH)2, activated Co(OH)2, and carbon paper. (c) TEM image, (d) SAED pattern, (e) HRTEM image of pristine Co(OH)2. (f) TEM image, (g, h) HR-TEM images selected in areas I and II, (i) STEM elemental mapping of Co(OH)2–60.
Figure 2
(a) iR-Corrected polarization curves in 1.0 mol/L KOH solution of various electrocatalysts as indicated (scan rate: 5 mV/s). (b) Tafel plots derived from the polarization curves in (a). (c) Chronopotentiometry curve of Co(OH)2 at a constant current density of −50 mA/cm2. (d) Nyquist plot measured at potential of −0.2 V vs. RHE.
Figure 3
(a) Co L-edge XAS and (b) Co K-edge XANES spectra of the pristine Co(OH)2 and Co(OH)2 after electrochemical activation. (c) In-situ Raman spectrum of Co(OH)2 under different activation stages. (d) Dark-field STEM image and (e) corresponding EELS mapping of Co(OH)2-60. (f) Dark-field STEM image and (g) corresponding EELS mapping of pristine Co(OH)2.

 DownLoad:
DownLoad:

 下载:
下载:





