Citation:
Hang Chen, Chengzhi Cui, Hebo Ye, Hanxun Zou, Lei You. Enhancing hydrolytic stability of dynamic imine bonds and polymers in acidic media with internal protecting groups[J]. Chinese Chemical Letters,
2024, 35(5): 109145.
doi:
10.1016/j.cclet.2023.109145
Enhancing hydrolytic stability of dynamic imine bonds and polymers in acidic media with internal protecting groups
English
Enhancing hydrolytic stability of dynamic imine bonds and polymers in acidic media with internal protecting groups
State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
b.
College of Environmental and Biological Engineering, Fujian Provincial Key Laboratory of Ecology-Toxicological Effects & Control for Emerging Contaminants, Putian University, Putian 351100, China
c.
College of Chemistry and Material Science, Fujian Normal University, Fuzhou 350007, China
d.
Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou 350108, China
* Corresponding authors at: State Key Laboratory of Structural Chemistry
Fujian Institute of Research on the Structure of Matter
lyou@fjirsm.ac.cn (L. You). 1 These authors contributed equally to this work.
Received Date:
29 May 2023 Accepted Date:
21 September 2023 Revised Date:
08 September 2023 Available Online:
15 May 2024
Abstract:
Imine bonds are among the most explored building motifs in dynamic chemistry, polymers, and materials, and yet, their acid-resistance remains a longstanding issue. Herein we demonstrate a concept of internal protecting groups for improving the kinetic stability of dynamic imine bonds and polymers. Systematic examination of structure-reactivity relationship of a series of aldehydes/imines bearing a neighboring carboxyl allowed uncovering of required structural features for dynamically masking imine bonds with cyclic structures. Mechanistic studies indicated that noncovalent interactions along with sterics control the ring-chain equilibrium and the stability of imine bonds. The incorporation of internal protecting groups into imine polymers further enabled their controlled stability in acidic media. Moreover, a combination of dynamic covalent network and coordination supramolecular network provided a facile means for the modulation of luminescent and mechanical properties of polymers. The strategies and results reported should be beneficial to molecular assemblies, dynamic polymers, biological delivery, and intelligent materials.
By combining the bonding strength of covalent bonds and reversibility of supramolecular interactions dynamic covalent chemistry (DCC) renders polymer species responsive and adaptable while maintaining the inherent stability [1-5]. The variation of the lifetime of dynamic covalent bonds offers ample opportunities for shaping polymer properties, including self-healing, shape-memory, and degradability/recyclability [6-10]. In particular, dynamic covalent polymer networks (DCPNs) [11-13], covalent adaptable networks (CANs) [14-16], or so-called vitrimers [17-19] have the dual advantage of the reprocessability of thermoplastics and the robustness of thermosets. Therefore, DCC has emerged as a powerful tool for the manipulation of polymer systems.
As the cornerstone of DCC [20-23], a variety of dynamic covalent reactions (DCRs) have been explored, proceeding via dissociative or associative exchange mechanism [24-26]. Being one of the most used DCRs the impact of imine bonds (C=N, Schiff base) reaches into the areas of both covalent polymers and discrete assemblies [27-30]. In addition to associative exchange via transimination or metathesis, imine bonds readily dissociate into carbonyls and primary amines under acidic conditions (Fig. 1a) [31-33]. Although such a feature allows pH-responsiveness and closed-loop degradation/recycling [34-36], the hydrolytic cleavage of imine bonds makes their polymers unstable for storage/use. A general approach for controlled stability and degradability when needed would be desired.
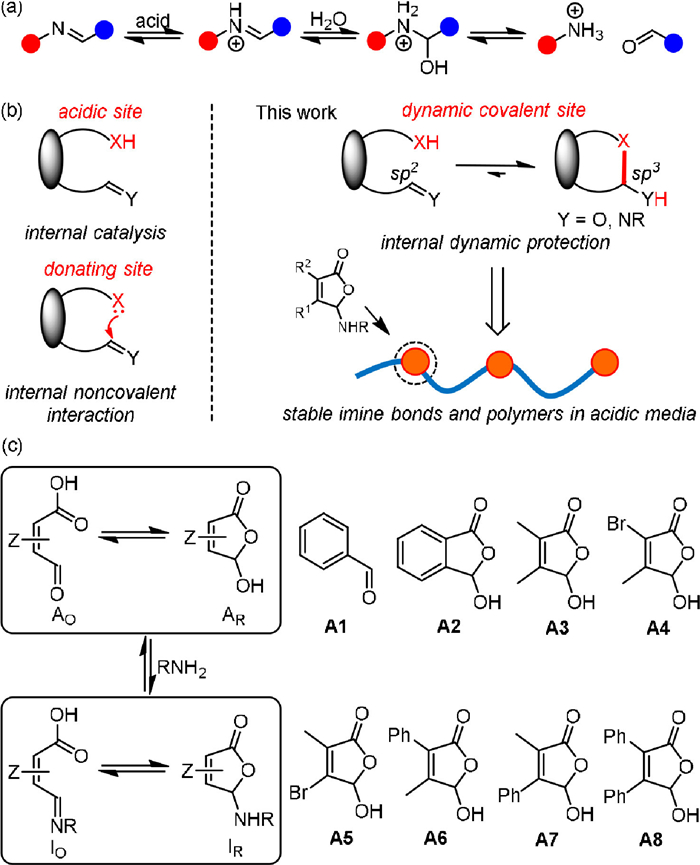
Figure 1
Figure 1.
(a) Acid-triggered hydrolysis of imine bond. (b) Illustration of internal catalysis and internal noncovalent interaction (left) and internal dynamic protecting groups toward acid-resistance of imine bonds and polymers (right). (c) Model ring-chain structures studied.
The concept of internal catalysis is generating intensive interest in dynamic covalent chemistry and polymers, as the neighboring participating group enables facile modulation of exchange kinetics (Fig. 1b) [37, 38]. Herein we demonstrate an internal "protecting" strategy to address the challenge of hydrolytic instability of imine bonds and polymer in acidic media (Fig. 1b). Building upon nearby assisting groups for thermodynamic stabilization through n→π* interactions (Fig. 1b) [39] and the control over dynamic covalent reactivity [40], the diverse scaffolds of vicinal aldehyde and carboxyl groups would offer the opportunity for masking the imine bonds in the form of ring tautomers (Fig. 1b). Nucleophilic attack on the tertiary hemiaminal ether carbon (sp3) would be kinetically disfavored over open imine (sp2) due to steric hindrance. The degree of ring-chain tautomerism would thus provide a handle for tuning hydrolytic stability of imines. In essence, the internal dynamic protecting group would be harnessed for the regulation of kinetic stability/degradability of imine bonds and their polymers.
With the strategy in place, a suite of model aldehydes was designed and prepared (Fig. 1c), including benzaldehyde (A1), 2-formylbenzoic acid (A2), and 1-carboxyl-2-formylethene derivatives (A3-A8) with varying substitution pattern, to unravel structural requirements for enhancing the stability of imines (Schemes S1-S3 in Supporting information). The ring tautomer of A2-A8 was dominant in CD3CN. Crystal structures of A3-A5 also supported the lactone form (CCDC number for A3-A5: 2255985-2255987, Fig. S31 in Supporting information).
The imines of aldehydes and 1-butylamine were then created in situ and isolated (I1-I8, Figs. S32–S43 in Supporting information). To probe the position of ring-chain tautomerization equilibrium, base titration was conducted in CD3CN. Although 2.0 equiv. of Et3N induced the formation of open aldehyde of A2, a large excess amount of Et3N was required with I2, suggesting enhanced preference of lactone structure for I2 over A2 (Figs. S44 and S45 in Supporting information). Furthermore, ring-opening was detected upon adding DBU to A3, but not Et3N (Figs. S46–S49 in Supporting information). Again more base was needed for I3 than A3 in order to obtain open aldehyde/imine. Similar findings were found with A7-A8 and their imines I7-I8 (Figs. S50–S53 in Supporting information). These results demonstrate that imines can be masked in a dynamic and tunable manner through delicate structural design.
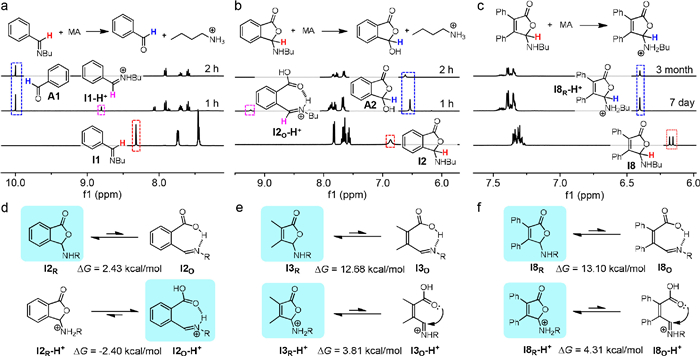
The hydrolytic stability of imines in acidic solution was next examined. After adding methanesulfonic acid (MA) I1 quickly (2 h) decomposed to recover A1, with iminium ion I1-H+ as the intermediate (Fig. 2a and Fig. S54 in Supporting information). The complete dissociation of I2 into A2 and 1-butylamine was also apparent with MA present after 2 h (Fig. 2b and Fig. S55 in Supporting information). Furthermore, open iminium ion (I2O-H+) emerged followed by the hydrolysis to create A2. In contrast, I3-I8 remained intact after 90 days upon treatment with MA (Figs. S56–S61 in Supporting information). There was a downfield shift from 6.20 ppm to 6.35 ppm for hemiaminal ether proton of I8 upon adding MA (Fig. 2c). Moreover, the imine survived even with D2O present. These observations fall in line with the protonation of cyclic hemiaminal ether nitrogen, protecting the imine from hydrolysis.
Figure 2
Figure 2.
Hydrolytic stability of imines with acid studied by experiments and calculations. 1H NMR spectra of I1 (a), I2 (b), and I8 (c) in CD3CN and changes with methanesulfonic acid (MA). Major structures and change in free energy (△G = GO – GR, O for form and R for ring form) by calculation for ring-chain equilibrium of I2 and I2-H+ (d), I3 and I3-H+ (e), and I8 and I8-H+ (f), with favored structures marked in blue.
To further elucidate the mechanism of the internal protecting group, DFT calculations were performed. The ring-chain tautomerization equilibrium of I2 favors the lactone form (I2R, Fig. 2d). This result might be attributed to n(N)→σ*(C-O) stabilizing interaction (i.e., anomeric effect) in the ring form over the hydrogen bond in the open isomer (I2O, Fig. S62 in Supporting information). The protonation of nitrogen lone pair sabotages n→σ* interaction and leads to a reversal of ring-chain equilibrium for I2-H+ (-2.40 kcal/mol, Fig. 2d and Fig. S63 in Supporting information). The planar structure of open iminium ion I2O-H+ would render it labile to hydrolysis, in agreement with experimental observations.
The preference of the cyclic hemiaminal ether was confirmed for I3 and I8 (12.68 and 13.10 kcal/mol, Figs. 2e and f). Compared to phenyl plane in I2, the ring tautomer for I3/I8 is highly favored due to the hindrance imposed by two methyl/phenyl groups on the olefin (Figs. S64 and S65 in Supporting information). Despite a dramatic drop of ΔG for I3-H+ and I8-H+ (3.81 and 4.31 kcal/mol), the domination of ring tautomer (I3R-H+ and I8R-H+) is maintained, confirming the crucial role of aforementioned steric effect. Two open isomers with carboxyl-imininium n→π* interaction account for the population (> 99%) for I3O-H+. Different from that of I2O-H+ (1.42 Å), the C=C bond within I3O-H+ has a much shorter bond distance (1.35 Å), causing the carboxyl to rotate to relieve the repulsion. The iminiuim unit remains nearly coplanar with olefin, engaging in n→π* interaction with the carboxyl. Analogous structural features were found for I8-H+ as I3-H+ (Figs. S66 and S67 in Supporting information). With the ring tautomer promoted by the carboxyl group, nucleophilic attack by water would be suppressed, supporting the strategy of internal protecting group.
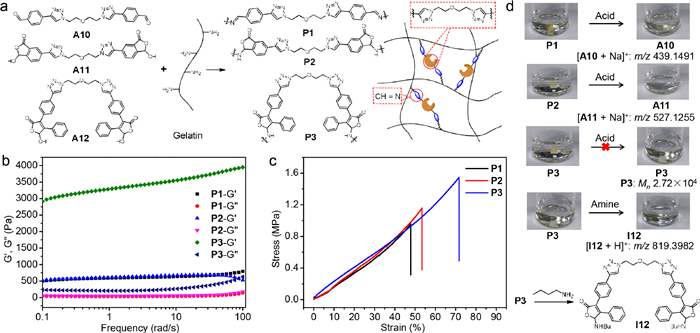
Having identified structural features for enhancing the stability of imines, dynamic covalent polymers were next constructed. Toward this end, three bifunctional aldehydes (A10-A12) with the identical flexible linkage were prepared (Fig. 2a and Schemes S4–S6 in Supporting information). Those aldehydes were then employed as cross-linkers for reactions with gelatin (i.e., a polyamine) in a mass ratio of 1:5 to create dynamic covalent polymer networks (hydrogel P1-P3) (Fig. 3a and Fig. S68 in Supporting information). Characterized by infrared spectroscopy (IR), the C=N vibration peak at 1608 cm−1 emerged for P1, while the vibration peak of C-N appeared at 1014 and 1033 cm−1 for P2 and P3, respectively (Figs. S69–S71 in Supporting information). These results supported the formation of open imine (P1) and cyclic hemiaminal ether (P2 and P3) linkages. The molecular weight values through gel permeation chromatography (GPC) are listed in Table S9 (Supporting information).
Figure 3
Figure 3.
Construction, characterization, and stability/degradability of dynamic covalent polymers. (a) Preparation of imine polymers P1-P3 from A10-A12 and gelation. (b) Oscillatory rheology analysis of P1-P3 through frequency sweep (1% strain). (c) Stress-strain curves of P1-P3 in tensile tests. (d) Degradation of P1-P3 in 1 mol/L HCl and P3 with 1-butylamine (3 mol/L) in H2O, with photographs and measured m/z or Mn shown.
Thermogravimetric analysis revealed the higher thermal stability of P3 than P1 and P2 (Fig. S72 in Supporting information). Glass transition temperature (Tg) values of −124.1, −123.2, and −113.5 ℃ for P1-P3 were obtained by differential scanning calorimetry, respectively (Fig. S73 in Supporting information). Such phenomena are likely due to the higher stability (i.e., strength) of hemiaminal ether linkage (C-O and C-N) than the imine bond (C=N). The sum of the bond dissociation energy of C-O (85-91 kcal/mol) and C-N (69-75 kcal/mol) is larger than that of C=N (143 kcal/mol), in accordance with the interpretation.
To investigate mechanical properties we performed viscoelasticity measurements through oscillatory rheology analysis (Fig. 3b). P1-P3 afforded a larger storage modulus (G′) than the loss modulus (G′′) at room temperature, demonstrating elastic solid feature of cross-linked hydrogels. In particular, the G′ value of P3 is 3-4 kPa, which is 6-8 folds over the storage modulus of P1 and P2 (~500 Pa). P1-P3 gave a fracture strength value of 0.95 MPa, 1.14 MPa, and 1.54 MPa in tensile tests. The elongation at break is 72% for P3, much larger than the value for P1 (48%) and P2 (53%). These results indicate the significantly higher mechanical strength for P3 than P1 and P2.
The hydrolytic stability of P1-P3 in acidic media was then studied (Fig. S74 in Supporting information). P1 completely decomposed and dissolved in 1 mol/L HCl after 30 min, and the formation of aldehyde monomers was validated in mass spectrometry (Fig. 3d and Fig. S75 in Supporting information). Analogous results were obtained with P2, which readily reversed into corresponding aldehydes and amines in acidic solution (Fig. 3d and Fig. S76 in Supporting information). The swelling of P3 was observed after placed in 1 mol/L HCl for 3 days, but no decomposition apparent (Fig. 3d and Fig. S77 in Supporting information). The molecular weight (M = 2.72 × 104, Đ = 1.82) remained nearly intact before and after acid treatment, confirming significantly improved acid-resistance for P3. The use of 5 mol/L HCl afforded similar results, with P3 survived and P1/P2 not. The strikingly different kinetic stability between P1-P3 is in agreement with model studies and showcases the capability of the internal protecting group for dictating the stability of imine polymers.
Having achieved hydrolytic stability of imine polymers under acidic condition, we then set to explore the degradability by taking advantage of DCC. Since the decomposition of P3via a dissociative mechanism is unlikely, the associative mechanism of transimination was utilized. P3 dissolved and degraded through amine exchange with 1-butylamine, as evidenced by 1-butylamine derived imine (I12) in mass spectrometry (Fig. 3d and Fig. S78 in Supporting information). By manipulating exchange mechanisms controlled stability and cleavability of imine polymers were thus attained.
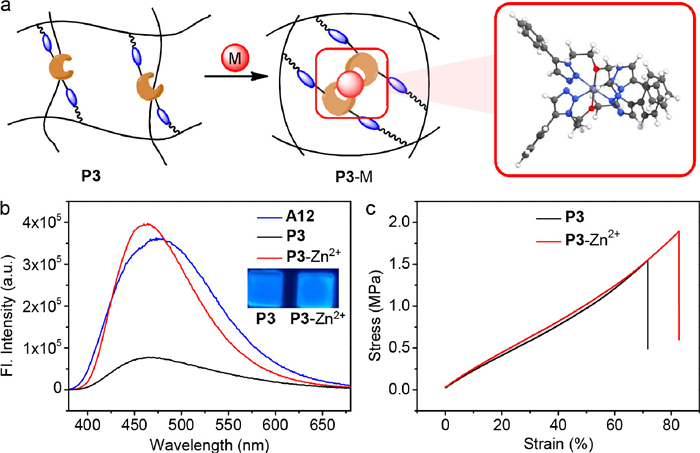
Finally, the impact of metal coordination sites on dynamic covalent polymer networks was studied, as the crosslinking via metal binding offers another handle for property modulation (Fig. 4a) [41-43]. Since diphenylethene is a part of aggregation-induced emission luminogen tetraphenylethene, [44-47] the luminescence of P3 was measured. The blue fluorescence of P3 was suppressed in solid-state emission spectra (Fig. 4b). However, when Zn(OTf)2 was present the emission was significantly turned on (Fig. 4b). Upon the crosslinking in the six-coordinated octahedral geometry of Zn2+ with triazole nitrogen and central oxygen atoms as binding sites the rotational freedom of the flexible linkages within P3 would be diminished (Fig. 4a and Fig. S80 in Supporting information). Furthermore, the rigidity improved by coordination leads to the inhibition of non-radiation pathway and the enhancement of the fluorescent quantum yield from 20% (P3) to 33% (P3-Zn2+) (Table S10 in Supporting information). Considering the Lewis acidity of zinc salts the stability of imine bonds was tested. The imine I12 from A12 and 1-butyamine remained intact in the presence of Zn(OTf)2 (Fig. S89 in Supporting information). Furthermore, metal coordination had an effect on enhancing the mechanical strength of polymers (Fig. 4c, Figs. S90 and S91 in Supporting information), as the energy from additional strain can be dissipated via the re-organization of supramolecular network [48-50].
Figure 4
Figure 4.
Illustration of a combination of covalent network and supramolecular network, with the proposed coordination mode calculated by DFT method (a), and the impact of metal binding on solid-state fluorescence (b) and stress-strain relationship (c) of P3.
In summary, we developed a general strategy of internal protecting groups for improving the stability of dynamic imine bonds and polymers under acidic condition. The structural features of nearby carboxyl induced aldehyde/imine ring-chain isomers governing the kinetic stability of imines were identified. Noncovalent interactions and steric hindrance play a crucial role in regulating the ring-chain equilibrium and stability of imine bonds. By incorporating internal protecting groups the controlled stability of imine polymer networks in acidic media was further achieved. Finally, a combination of covalent network and coordination supramolecular network allowed facile regulation of fluorescent and mechanical properties of polymers. The results reported should find applications in dynamic assemblies, adaptable polymers, and smart materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank the National Natural Science Foundation of China (NSFC, Nos. 22071247, 92156010, 22101283, and 22101284), the Key Research Program of Frontier Sciences (No. QYZDBSSW-SLH030) of the CAS, Natural Science Foundation of Fujian Province (Nos. 2020J06035 and 2022J05085), and Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (No. 2021ZR112) for support.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109145.
[1]
N. Roy, B. Bruchmann, J.M. Lehn, Chem. Soc. Rev. 44 (2015) 3786–3807. doi: 10.1039/C5CS00194C
[2]
P. Chakma, D. Konkolewicz, Angew. Chem. Int. Ed. 58 (2019) 9682–9695. doi: 10.1002/anie.201813525
G.R. Whittell, M.D. Hager, U.S. Schubert, I. Manners, Nat. Mater. 10 (2011) 176–188. doi: 10.1038/nmat2966
[50]
L.J. Chen, G.Z. Zhao, B. Jiang, et al., J. Am. Chem. Soc. 136 (2014) 5593–6001.
Figure 1
(a) Acid-triggered hydrolysis of imine bond. (b) Illustration of internal catalysis and internal noncovalent interaction (left) and internal dynamic protecting groups toward acid-resistance of imine bonds and polymers (right). (c) Model ring-chain structures studied.
Figure 2
Hydrolytic stability of imines with acid studied by experiments and calculations. 1H NMR spectra of I1 (a), I2 (b), and I8 (c) in CD3CN and changes with methanesulfonic acid (MA). Major structures and change in free energy (△G = GO – GR, O for form and R for ring form) by calculation for ring-chain equilibrium of I2 and I2-H+ (d), I3 and I3-H+ (e), and I8 and I8-H+ (f), with favored structures marked in blue.
Figure 3
Construction, characterization, and stability/degradability of dynamic covalent polymers. (a) Preparation of imine polymers P1-P3 from A10-A12 and gelation. (b) Oscillatory rheology analysis of P1-P3 through frequency sweep (1% strain). (c) Stress-strain curves of P1-P3 in tensile tests. (d) Degradation of P1-P3 in 1 mol/L HCl and P3 with 1-butylamine (3 mol/L) in H2O, with photographs and measured m/z or Mn shown.
Figure 4
Illustration of a combination of covalent network and supramolecular network, with the proposed coordination mode calculated by DFT method (a), and the impact of metal binding on solid-state fluorescence (b) and stress-strain relationship (c) of P3.

 DownLoad:
DownLoad:

 下载:
下载:





