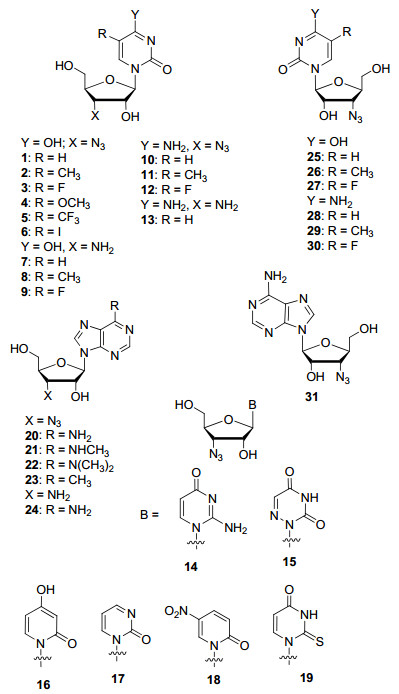
 Figure1.
3'-Azido-3'-deoxy-D/L-nucleosides
Figure1.
3'-Azido-3'-deoxy-D/L-nucleosides
Citation:
Ren Hang, Tao Jingchao, An Haoyun. Synthesis of 3'-Azido-D/L-nucleosides[J]. Chinese Journal of Organic Chemistry,
2018, 38(1): 138-147.
doi:
10.6023/cjoc201708005

3'-叠氮D/L-核苷的合成
摘要:
报道了两种正交保护的3'-叠氮-3'-脱氧-D/L-核糖关键中间体的合成,此二关键中间体与各种嘧啶、吡啶、嘌呤相关的杂环碱基进行糖苷化,得到了相应的3'-叠氮-3'-脱氧D核苷衍生物以及3'-叠氮-3'-脱氧L核苷衍生物.合成了药物相关的3'-叠氮-3'-脱氧-6-氮杂尿苷、4-脱氧尿苷、2-硫代尿苷、3-去氮尿苷、硝基吡啶核苷、异胞苷衍生物.31个最终产品中的14个是新化合物,其结构均得以确证.所有最终产品都是用本文描述的平行法从相应的新关键中间体合成而得.此二关键中间体可以用于合成含有任何不同碱基的新型核苷衍生物.
English
Synthesis of 3'-Azido-D/L-nucleosides
Abstract:
Two orthorganally protected 3'-azido-3'-deoxy-D/L-ribosides were synthesized and successfully glycosylated with various pyrimidine, pyridine and purine related heterocyclic bases. 3'-Azido-3'-deoxy-D-pyrimidine nucleosides, purine nucleosides as well as 3'-azido-3'-deoxy-L-nucleosides were synthesized. 3'-Azido-3'-deoxy-6-azauridine, 4-deoxyuridine, 2-thiouridine, 3-deazauridine, nitropyridinone and isocytidine derivatives were also synthesized as the drug analogues to explore their biological properties. 14 final products are novel and well characterized, while all of the 31 final products were synthesized by the new strategy from the corresponding novel key intermediates. The two key intermediates can be utilized to make all kinds of novel nucleoside derivatives by glycosylating with various heterocyclic bases.
-
Key words:
- azido-nucleoside
- / L-nucleoside
- / D-nucleoside
- / synthesis
- / glycosylation
-
1 Introduction
A number of nucleoside anticancer and antiviral drugs play an important role in human health.[1] Azido-modified nucleosides, such as zidovudine, are efficient reverse transcriptase (RT) inhibitors and have been used as efficient antiviral agents.[2] A recently discovered azido-nucleoside drug, azvudine, [3] is in clinical studies for the treatment of human immunodeficiency virus (HIV) and demonstrates activity against other viruses. Therefore, more azido-modified nucleoside drugs shall be identified to address long running unmet medical needs. Due to synthetic challenges of azido-modified nucleosides, the true scope of their biological activity has not been thoroughly investigated.[4] In addition, these azido group-containing molecules and their corresponding amino group-containing derivatives can be efficiently utilized for further conjugation, and in return, the conjugated molecules[5] can be used to accelerate antisense, small RNA and genomic related technologies and further advance of modern diagnostic application.
To further expand the application of small molecule and genomic drug discovery, it is essential to explore the synthesis of azido-modified nucleosides. Herein, we report the efficient synthesis of 3'-azido-modified D-nucleosides (compounds 1~24, Figure 1) and L-nucleosides (compounds 25~31, Figure 1). To avoid tedious synthesis starting from nucleosides, we utilized readily available D-and L-azido-modified ribosides 32 and 43 as starting materials.
Figure1.
3'-Azido-3'-deoxy-D/L-nucleosides
2 Results and discussion
Some 3-azido-and amino-nucleosides were synthesized by traditional methods starting from nucleosides.[6] The methods do not allow the late-stage functionalization for the synthesis of a structurally diverse nucleoside library. Therefore, we decided to utilize a well-protected 3'-azido-3'-deoxyriboside which can be glycosylated with any heterocyclic base, enabling the synthesis of all possible azido-nucleoside derivatives. The reported literature for the synthesis of benzoyl and acetyl protected 3'-azido-3'-deoxy-riboside suffer from the formation of the elimination side product in over 40% in the azido nucleophilic substitution step.[6, 7] We constructed 1, 2-di-O-acetyl-5-O-(4-methyl-benzoyl)-D-riboside (32) (Scheme 1) in large scale from D-(+)-xylose with less than 20% eliminated side product in the key step. The developed process for the synthesis of fully-protected 3'-azido-riboside 32 was also adopted for the synthesis of new 1, 2-di-O-acetyl-5-O-(4-methylbenzoyl)-L-riboside (43). With these two orthogonally protected ribosides in hands, we were able to synthesize various new azido-modified nucleosides.
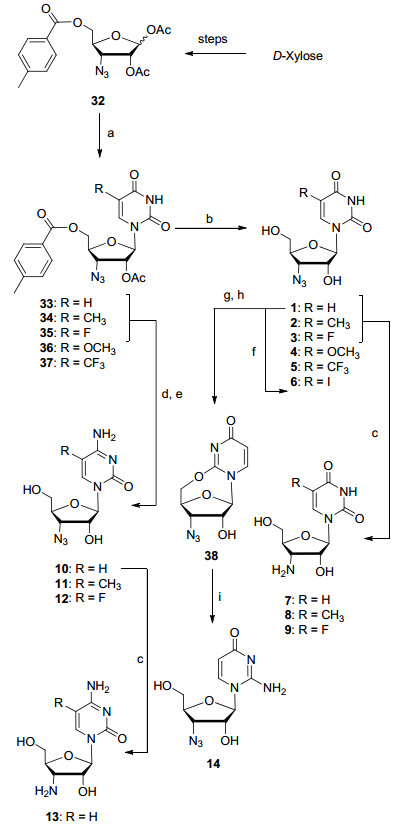 Scheme1.
Synthesis of 3'-azido-3'-deoxy-D-pyrimidine nucleosides 1~14
Scheme1.
Synthesis of 3'-azido-3'-deoxy-D-pyrimidine nucleosides 1~14
The orthogonally protected 3'-azido-3'-deoxyriboside 32 was glycosylated with uracil, 5-methyluracil, 4-fluoro-uracil, 5-methoxyuracil and 5-trifluoromethyl uracil under TMS-Tf conditions to provide the corresponding protected nucleosides 33~37. They were deprotected under saturated ammonia solution in methanol to give the desired uridine and 5-substituted uridine derivatives 1~5. 3'-Azido-3'-deoxyuridine (1) was iodinated, resulting in the formation of 5-iodouridine derivative 6. Compounds 1~3 were hydrogenated with Pd/C as the catalyst to generate desired 3'-amino-3'-deoxyuridine derivatives 7~9. The protected key intermediates 33~35 (R=H, CH3 and F) were converted to the corresponding cytidine derivatives 10~12 utilizing POCl3/triazole method.[8] The 3'-azido-3'-deoxy-cytidine (10) was hydrogenated to 3'-amino-3'-deoxy-cytidine (13). 3'-Azido-3'-deoxyuridine (1) reacted with p-methylphenylsulfonyl chloride to generate the 5'-tosylate, which was cyclized with the oxygen atom at the 2-position on the uracil ring to form 2, 5'-anhydro-uridine 38. Uridine 38 was then treated with ammonium hydroxide to complete the synthesis of 3'-azido-3'-deoxy-isocytidine (14).
6-Azauridine and 2', 3', 5'-tri-O-acetyl-6-azauridine, aza-ribine, are all antimetabolic antineoplastic agents.[9] The synthase inhibitor 3-deazauridine[10] and the DNA methylation inhibitor 4-deoxyuridine[11] have been used as anticancer drugs. 2-Thiouridine and 5-nitropyridone ribofuranosyl nucleosides are also biologically relevant molecules. In order to explore the biological properties of these drugs with substitution at the 3'-position with an azide, we synthesized the 3'-azido-3'-deoxy derivatives of these drugs. The orthogonally protected azido-riboside 32 was glycosylated with 6-azauracil, and the resulting compound was deprotected with saturated ammonia in methanol to furnish 3'-azido-modified 6-azauridine analogue 15. Riboside 32 was glycosylated with 2, 6-dihydroxypyridine and 2-hy-droxypyrimidine. The resulting protected nucleoside intermediates were deprotected to produce 3'-azido-3'-deoxy-3-deazauridine (16) and 3'-azido-3'-deoxy-4-deoxyuridine (17), which are the 3'-azido-modified derivatives of 3-dezazurine and 4-deoxyuridine drugs. The same glycosylation and deprotection strategy were used for the synthesis of compounds 18 and 19 where riboside 32 was glycosylated with 5-nitro-pyridin-2-one and 2-thiouracil (Scheme 2).
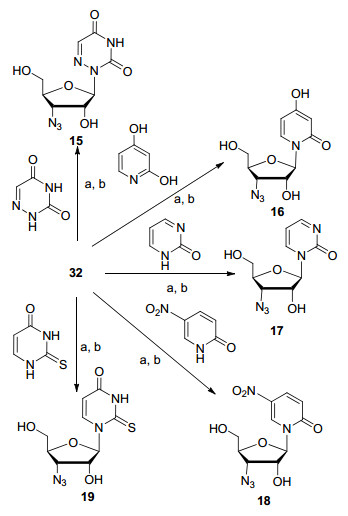 Scheme2.
Synthesis of 3'-azido-3'-deoxy-D-nucleosides 15~19 with special heterocyclic bases
Scheme2.
Synthesis of 3'-azido-3'-deoxy-D-nucleosides 15~19 with special heterocyclic bases
3'-Azido-3'-deoxyadenosine (20) and 3'-amino-3'-deoxy-adenosines (24) were previously synthesized from adenosine or xylo-adenosine[4] using a long synthetic route that required functional groups manipulation and hydroxyl group α/β-conversion. In addition, this strategy does not enable late-stage functionalization to install various heterocycles. Robins and co-workers[12] also converted 3'-azido-adenosine to 6-dialkylsubstituted derivatives by the triazole strategy which could be much easier if chloro-is at the 6-position of such adenosine analogue, such as intermediate 39 (Scheme 3). We glycosylated riboside 32 with 6-methylpurine, and the resulted compound was deprotected with saturated ammonia in methanol resulting in the desired 3'-aziso-3'-deoxy-6-methylpurine nucleoside 23 which is the derivative of natural product 6-methylpurine riboside. Riboside 32 was also glycosylated with 6-chloro-adenine resulting a versatile intermediate 39, which can be utilized to make all kinds of 6-modified purine nucleoside derivatives.
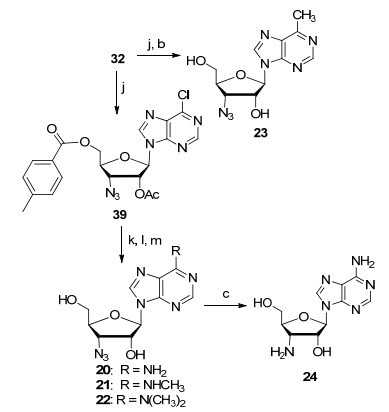 Scheme3.
Synthesis of 3'-azido-3'-deoxy-D-purine nucleosides 20~24
Scheme3.
Synthesis of 3'-azido-3'-deoxy-D-purine nucleosides 20~24
The key intermediate 39 was synthesized and treated with ammonia resulting in desired compound 20. Compound 39 was also treated with methylamine and N, N-dimethylamine. The chloro at position 6 was replaced and the protecting groups were removed at the same time to furnish the synthesis of N6-methylated compounds 21 and 22. Hydrogenation of compound 20 gave 3'-amino-3'-deoxyadenosine 24. Compounds 20 and 24 were synthesized by Kim and co-workers[13] with long route and OH conversion starting from adenosine which can only be used for the synthesis of this compound. Therefore, the key intermediate 39 can be used as a versatile scaffold to make all kinds of 6-N-substituted and C-substituted adenosine derivatives by nucleophilic substitution and various C—C bond formation reactions. In addition, the amino and azido groups on the derivatives can be used for various conjugation.
L-Nucleosides demonstrate special biological property and have different application compared to the normal D-nucleosides.[14] With the developed protocol for the synthesis of the protected 3'-azido-3'-deoxyriboside 32 ready at Granlen, we transferred the strategy to the synthesis of new 1, 2-di-O-acetyl-5-O-(4-methylbenzoyl)-L-riboside (43) (Scheme 4). 1, 2-O-Isopropylidene-α-L-xylofuranose (40) was synthesized from L-(-)-xylose according to the reported protocol.[15] Compound 40 was selectively protected and then activated with Tf2O giving the active triflate intermediate 41. Further nucleophilic substitution with sodium azide resulted in the 3'-azido-L-riboside 42. Further treatment with acetic anhydride under acidic condition by our routine protocol furnished the synthesis of 3'-azido-3'-deoxy-L-riboside 43.
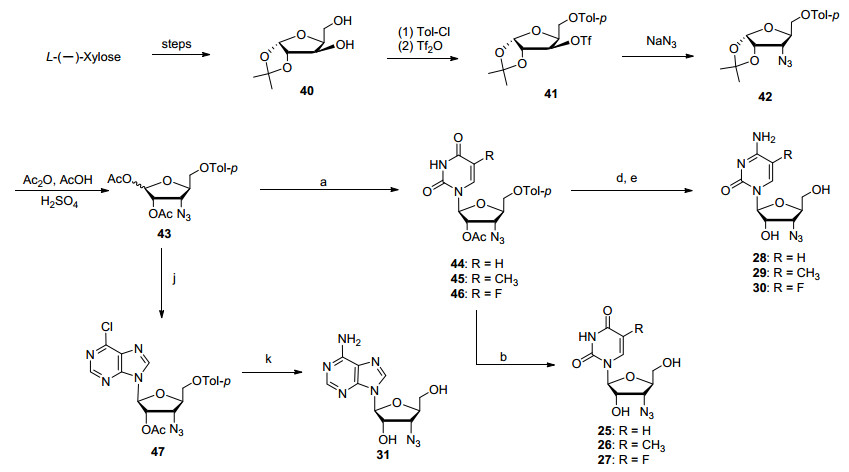 Scheme4.
Synthesis of 3'-azido-3'-deoxy-L-riboside 43 and nucleosides 25~31
Scheme4.
Synthesis of 3'-azido-3'-deoxy-L-riboside 43 and nucleosides 25~31
The key intermediate 3'-azido-3'-deoxy-L-riboside 43 was glycosylated with uracil, 5-methyluracil and 5-fluoro-uracil. The resulting intermediates 44~46 were deprotected with ammonia to give the desired 3'-azido-3'-deoxy-L-uridine derivatives 25~27. The key intermediates 44~46 were reacted with POCl3/triazole, followed by the treatment with ammonia, giving the desired 3'-azido-3'-deoxy-L-cytidine derivatives 28~30, respectively. L-Riboside 43 was glycosylated with 6-chloropurine, and the key intermediate 47 was obtained. It was further treated with ammonia giving the desired 3'-aziso-3'-deoxy-L-adenosine (31). Therefore, our parallel-last strategy just stated starting from the versatile D-and L-ribosides 32 and 43 can be utilized broadly to make a variety of L-nucleosides with all kinds of heterocyclic bases, and more derivatives can be made from the key intermediates (Scheme 4).
3 Conclusions
In conclusion, we have developed the protocol and synthesized the fully-protected 3'-azido-3'-deoxy-D-riboside 32 in large scale, and synthesized new protected 3'-azido-3'-deoxy-L-riboside 43. These D-and L-riboside key intermediates were successfully glycosylated with various substituted uracil, adenine, 6-azauracil, 2-thiouracil, pyrimidone, and pyridinone related heterocyclic bases in a parallel fashion for the synthesis of the diversified nucleoside derivatives after deprotection and simple functional group manipulation. 3'-Azido-3'-deoxy-D-nucleoside derivatives 1~6, 10~12 and 14~23 and L-nucleoside derivatives 25~31 were obtained and ready for further biological and genomic studies. The key riboside intermediates 32 and 43 reported here can be utilized further for the synthesis of various novel nucleoside derivatives with other heterocyclic bases to expand further research in the related fields.
4 Experimental
4.1 Instrument and reagent
Starting materials and reagents were obtained from commercial suppliers and were used without further purification unless otherwise stated. 1H NMR spectra were obtained with a Bruker Avance 400 spectrometer using DMSO-d6 downfield with respect to an internal δ as solvents. Chemical shifts are reported as standard of tetramethylsilane (TMS). Product purity was tested by an Agilent 1260 analytical HPLC system. LC-MS spectra were measured on an Agilent 6120 LC-MS spectrometer. TLC was performed on silica gel GF254. Flash chromatography was performed on silica gel 200~300 mesh (Yantai Silica Gel Co. LTD). 1, 2-Di-O-acetyl-5-O-(4-methylbenzoyl)-D-ribo-side (32) was prepared from D-xylose by adopting literature strategy with modification and different protecting scheme.[7] 6-Methylpurine was prepared from 6-chloro-purine according to procedures described in the literature.[16]
4.2 Experimental procedures
4.2.1 General procedure for the glycosylation of uracil, 5-methyluracil, 4-fluorouracil, 5-methoxyuracil, and 5-tri-fluoromethyluracil with 1, 2-di-O-acetyl-5-O-(4-methyl-benzoyl)riboside (32)
To a suspension of uracil, 5-methyluracil, 4-fluorouracil, 5-methoxyuracil or 5-trifluoromethyluracil (9.6 mmoL, 1.2 equiv.) in anhydrous acetonitrile (100 mL) was added N, O-bis(trimethylsilyl)acetamide (7.8 mL, 31.8 mmol, 4.0 equiv.) dropwise with stirring. The mixture was stirred for 1 h at room temperature. Then 1, 2-di-O-acetyl-5-O-(4-methyl benzoyl)riboside (32, 3.0 g, 7.95 mmol, 1.0 equiv.) was added, and the reaction mixture was cooled to 0 ℃ followed by addition of trimethylsilyl triflate (4.30 mL, 23.85 mmol, 3.0 equiv.). The mixture was stirred overnight at room temperature. Upon completion of the reaction as monitored by thin-layer chromatography (TLC), the reaction mixture was poured into ice water and then treated with a mixture of saturated sodium bicarbonate and ethyl acetate. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column using petroleum ether-ethyl acetate as eluents providing products 33~37 as white solids in 82%~93% yields.
4.2.2 General procedure for the conversion of compounds 33~37 to compounds 1~5
The obtained compounds 33~37 were dissolved in NH3-MeOH, and the reaction mixture was allowed to stir at room temperature overnight. Upon completion of the reaction as monitored by TLC, the solution was concentrated in vacuum under reduced pressure, and the residue was purified on a silica gel column resulting in compounds 1~5 in 78%~95% yields as white solids.
3'-Azido-3'-deoxyuridine (1): gray solid, 76% yield in two steps with an HPLC purity of 98.7%. Rf=0.4 (dichloromethane/methanol, V: V=10: 1). m.p. 130~135 ℃ (dec.); 1H NMR (DMSO-d6, 400 MHz) δ: 3.48~3.70 (m, 2H, CH2), 3.80~4.10 (m, 2H, 2', 3'-H), 4.42 (m, 1H, 4'-H), 5.28 (br, 1H, 5'-OH), 5.66 (d, J=8.0 Hz, 1H, ArH), 5.76 (d, J=5.2 Hz, 1H, 2'-OH), 6.17 (s, 1H, 1'-H), 7.87 (d, J=8.4 Hz, 1H, ArH), 11.29 (s, 1H, NH); ESI-MS m/z: 269.9 [M+H]+, 291.9 [M+Na]+.
3'-Azido-3'-deoxy-5-methyl uridine (2):[17] light yellow foam, 77% yield in two steps with an HPLC purity of 99.8%. Rf=0.6 (dichloromethane/methanol, V: V=10: 1). ESI-MS m/z: 284.0 [M+H]+, 305.9 [M+Na]+.
3'-Azido-3'-deoxy-5-fluorouridine (3): light yellow foam, 73% yield in two steps with an HPLC purity of 97.7%. Rf=0.2 (petroleum ether-ethyl acetate, V: V=1: 1). 1H NMR (DMSO-d6, 400 MHz) δ: 3.55~3.75 (m, 2H, CH2), 3.91~4.07 (m, 2H, 3, 4'-H), 4.37~4.45 (m, 1H, 2'-H), 5.42 (br, 1H, 5'-OH), 5.70~5.75 (m, 1H, 2'-OH), 6.18 (d, J=5.2 Hz, 1H, 1'-H), 8.27 (d, J=7.2 Hz, 1H, ArH), 11.62 (s, 1H, NH); ESI-MS m/z: 287.8 [M+H]+.
3'-Azido-3'-deoxy-5-methoxy uridine (4): light yellow solid, 72% yield over two steps with an HPLC purity of 95.7%. Rf=0.7 (dichloromethane/methanol, V: V=10: 1). m.p. 150~155 ℃ (dec.); 1H NMR (DMSO-d6, 400 MHz) δ: 3.56~3.64 (m, 4H, CH3, CH), 3.66~3.78 (m, 1H, CH), 3.90~3.98 (m, 1H, 4'-H), 4.08 (t, J=5.2 Hz, 1H, 3'-H), 4.42~4.52 (m, 1H, 2'-H), 5.42 (t, J=4.4 Hz, 1H, 5'-OH), 5.79 (d, J=4.8 Hz, 1H, 2'-OH), 6.14 (d, J=5.6 Hz, 1H, 1'-H), 7.61 (s, 1H, ArH), 11.48 (s, 1H, NH); ESI-MS m/z: 299.8 [M+H]+, 322.7 [M+Na]+; HRMS calcd for C10H13N5NaO6 [M+Na]+ 322.0764, found 322.0761.
3'-Azido-3'-deoxy-5-trifluoro methyluridine (5): yellow foam, 65% yield over two steps with an HPLC purity of 95.8%. Rf=0.4 (dichloromethane/methanol, V: V=15: 1). 1H NMR (DMSO-d6, 400 MHz) δ: 3.50~3.85 (m, 2H, CH2), 3.90~4.10 (m, 2H, 4'-H, 3'-H), 4.40~4.50 (m, 1H, 2'-H), 5.53 (t, J=4.4 Hz, 1H, 5'-OH), 5.69 (d, J=2.4 Hz, 1H, 2'-OH), 6.28 (d, J=4.8 Hz, 1H, 1'-H), 8.83 (s, 1H, ArH), 11.41 (s, 1H, NH); ESI-MS m/z: 359.7 [M+Na]+.
4.2.3 Synthesis of 3'-azido-3'-deoxy-5-iodouridine (6)
Iodine monochloride (2.4 g, 14.8 mmol, 2.0 equiv.) was added to a suspension of sodium azide (1.4 g, 22.2 mmol, 3.0 equiv.) in acetonitrile (50 mL) at ice-bath temperature with stirring. This mixture was stirred for another 5 min, and a solution of 3'-azido-3'-deoxyuridine 1 (2.0 g, 7.4 mmol) in acetonitrile (60 mL) was then added. The resulting reaction mixture was warmed to 25 ℃ and stirred for 24 h. Upon completion of the reaction as monitored by TLC, solvent was evaporated and the crude product was purified by flash chromatography on a silica gel column using dichloromethane-methanol as eluent to afford 1.5 g compound 6 as a white solid in 70% yield with an HPLC purity of 97.2%. Rf=0.4 (dichloromethane/methanol, V: V=10: 1). m.p. 170~175 ℃ (dec.); UV-vis (MeOH) λmax: 285 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 3.55~3.75 (m, 2H, CH2), 3.92~3.97 (m, 1H, 4'-H), 4.03 (t, J=5.2 Hz, 1H, 3'-H), 4.41~4.46 (m, 1H, 2'-H), 5.43 (t, J=4.8 Hz, 1H, 5'-OH), 5.70 (d, J=4.4 Hz, 1H, 2'-OH), 6.17 (d, J=5.2 Hz, 1H, 1'-H), 8.45 (s, 1H, ArH), 11.72 (s, 1H, NH); ESI-MS m/z: 395.7 [M+H]+, 418.6 [M+Na]+; HRMS calcd for C9H10IN5NaO5 [M+Na]+ 417.9624, found 417.9627.
4.2.4 General procedure for the synthesis of compounds 7~9
Compound 1 or 2 or 3 was dissolved in anhydrous methanol and triethylamine (1.0 equiv.). Catalyst 10% Pd/C (50% w/w) was added, and the mixture was stirred for 5 h under H2 (344.7 kPa) atmosphere. Upon completion of the reaction as monitored by TLC, the catalyst was filtered off through Celite. The solvent was evaporated and the crude product was purified by flash chromatography on a silica gel column using dichloromethane-methanol as eluent giving compounds 7~9, respectively, in 80%~86% yields as white solids.
3'-Amino-3'-deoxyuridine (7):[18] white foam, 86% yield with an HPLC purity of 96.0%. Rf=0.30 (dichloromethane/methanol, V: V=5: 1). UV-vis (MeOH) λmax: 265 nm; ESI-MS m/z: 244.0 [M+H]+.
3'-Amino-3'-deoxy-5-methyl uridine (8):[17] white foam, 83% yield with an HPLC purity of 96.6%. Rf=0.40 (dichloromethane/methanol, V: V=5: 1). UV-vis (MeOH) λmax: 268 nm; ESI-MS m/z: 258.0 [M+H]+.
3'-Amino-3'-deoxy-5-fluoro uridine (9):[19] white solid, 80% yield with an HPLC purity of 95.0%. Rf=0.45 (dichloromethane/methanol, V: V=5: 1). UV-vis (MeOH) λmax: 270 nm; ESI-MS m/z: 261.9 [M+H]+.
4.2.5 General procedure for the synthesis of 3'-azido-3'-deoxycytidine derivatives 10~12
To a solution of 1, 2, 4-triazole (5 equiv.) in dry dichloromethane was added phosphorus oxychloride (3.0 equiv.) followed by triethylamine (3.0 equiv.) at 0 ℃ and the reaction mixture was stirred for 30 min at 0 ℃. A solution of compound 33 or 34 or 35 (1.0 equiv.) in dry dichloromethane was then added dropwise. The mixture was moved to room temperature and was stirred for 1 h. Upon completion of the reaction as monitored by TLC, the reaction mixture was poured into ice water and was treated with saturated sodium bicarbonate and ethyl acetate. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated under reduced pressure. The residue was dissolved in NH3-MeOH, and the reaction mixture was allowed to stir at room temperature overnight. Upon completion of the reaction as monitored by TLC, the solution was concentrated in vacuum under reduced pressure, and the residue was purified on a silica gel column resulting in compounds 10~12, respectively, in 76%~85% yields as white solids.
3'-Azido-3'-deoxycytidine (10):[20] light yellow solid, 85% yield with an HPLC purity of 98.2%. Rf=0.2 (dichloromethane/methanol, V: V=5: 1). UV-vis (MeOH) λmax: 270 nm; ESI-MS m/z: 268.8 [M+H]+.
3'-Azido-3'-deoxy-5-methyl cytidine (11): white foam, 76% yield in two steps with an HPLC purity of 98.3%. Rf=0.45 (dichloromethane/methanol, V: V=5: 1). UV-vis (MeOH) λmax: 280 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 1.82 (s, 3H, CH3), 3.50~3.72 (m, 2H, CH2), 3.84~3.92 (m, 1H, 4'-H), 3.97 (t, J=5.2 Hz, 1H, 3'-H), 4.32~4.40 (m, 1H, 2'-H), 5.25 (t, J=5.2 Hz, 1H, 5'-OH), 5.77 (d, J=4.8 Hz, 1H, 2'-OH), 6.07 (d, J=5.2 Hz, 1H, 1'-H), 6.84 (s, 1H, NH), 7.33 (s, 1H, NH), 7.64 (s, 1H, ArH); ESI-MS m/z: 383.3 [M+H]+, 565.5 [2M+H]+.
3'-Azido-3'-deoxy-5-fluorocytidine (12): white solid, 79% yield in two steps with an HPLC purity of 99.0%. Rf=0.45 (dichloromethane/methanol, V: V=5: 1). m.p. 165~170 ℃ (dec.); UV-vis (MeOH) λmax: 239, 280 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 3.54~3.61 (m, 1H, CH), 3.69~3.76 (m, 1H, CH), 3.90~3.98 (m, 2H, 3'-H, 4'-H), 4.30~4.36 (m, 1H, 2'-H), 5.37 (t, J=5.2 Hz, 1H, 5'-OH), 5.70~5.74 (m, 1H, 2'-OH), 6.15 (d, J=5.2 Hz, 1H, 1'-H), 7.54 (s, 1H, NH), 7.78 (s, 1H, NH), 8.16 (d, J=7.2 Hz, 1H, ArH); ESI-MS m/z: 287.1 [M+H]+, 309.1 [M+Na]+, 325.0 [M+K]+. HRMS calcd for C9H11FN6NaO4 [M+ Na]+ 309.0724, found 309.0725.
4.2.6 Synthesis of 3'-amino-3'-deoxycytidine (13)
This compound was synthesized as described in Scheme 1 from compound 10 (2.0 g, 7.5 mmoL). 1.4 g of compound 13[19] was obtained as a white foam in 75% yield with an HPLC purity of 97.3%; Rf=0.30 (dichloromethane/ methanol, V: V=2: 1). UV-vis (MeOH) λmax: 270 nm; ESI-MS m/z: 243.0 [M+H]+.
4.2.7 Synthesis of 3'-deoxy-3'-azido-isocytidine (14)
To a solution of 3'-azido-3'-deoxyuridine (1) (5.0 g, 18.6 mmol) in dry pyridine (70 mL) at 0 ℃ with stirring was added p-toluenesulfonyl chloride (4.3 g, 22.32 mmol, 1.2 equiv.). The reaction mixture was stirred overnight at room temperature. Upon completion of the reaction as monitored by TLC, the reaction mixture was poured into ice water and was treated with saturated sodium bicarbonate and ethyl acetate. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated under reduced pressure. The residue was then dissolved in acetonitrile (100 mL), and 1, 8-diazabicyclo-[5.4.0]undec-7-ene (5.6 mL, 37.2 mmol, 2.0 equiv.) was added to the solution. The reaction mixture was heated to reflux for 1 h. Upon completion of the reaction as monitored by TLC, the hot solution was immediately filtered through a Celite pad, and the filtrate was evaporated to dryness. The residue was purified on a silica gel column to provide 3.0 g of product 38. The obtained compound 38 was dissolved in 30 mL of MeOH, and 30 mL of ammonium hydroxide was added. The reaction mixture was allowed to stir at room temperature overnight. Upon completion of the reaction as monitored by TLC, the solution was concentrated in vacuum under reduced pressure, and the residue was purified on a silica gel column resulting in 2.6 g of compound 14 as a white solid in 52% yield for three steps with an HPLC purity of 95.8%. Rf=0.2 (dichloromethane/methanol, V: V=5: 1). m.p. 164~169 ℃ (dec.). 1H NMR (DMSO-d6, 400 MHz) δ: 3.62~3.70 (m, 3H, 4'-H, CH2), 4.02 (t, J=2.8 Hz, 1H, 3'-H), 4.30~4.45 (m, 1H, 2'-H), 5.35 (t, J=4.8 Hz, 1H, 5'-OH), 5.54 (d, J=7.6 Hz, 1H, ArH), 5.76 (d, J=6.0 Hz, 1H, 2'-OH), 6.17 (d, J=4.8 Hz, 1H, 1'-H), 6.84 (s, 2H, NH2), 7.61 (d, J=8.0 Hz, 1H, ArH); ESI-MS m/z: 269.1 [M+H]+. HRMS calcd for C9H12N6NaO4 [M+Na]+ 291.0818, found 291.0815.
4.2.8 Synthesis of 3'-azido-3'-deoxy-D-nucleosides 15~19
These compounds were synthesized by the glycosylation of 6-azauracil, 2, 6-dihydroxypyridine, 2-hydroxypyrimi-dine, 5-nitro-pyridin-2-one and 2-thiouracil with 32. The resulted compounds were further deprotected resulted in final products 15~19.
3'-Azido-3'-deoxy-6-azauridine (15): yellow foam, 71% yield in two steps with an HPLC purity of 98.3%. Rf=0.5 (dichloromethane/methanol, V: V=10: 1). UV-Vis (MeOH) λmax: 262 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 3.40~3.60 (m, 3H, CH2, 4'-H), 3.88~4.00 (m, 2H, 2', 3'-H), 4.54~4.58 (m, 1H, 5'-OH), 4.85 (br, 1H, 2'-OH), 5.91 (d, 1H, J=3.6 Hz, 1'-H), 6.10 (br, 1H, NH), 7.53 (s, 1H, ArH); ESI-MS m/z: 271.1 [M+H]+, 293.0 [M+Na]+.
3'-Azido-3'-deoxy-3-deaza uridine (16): white foam, 68% yield over two steps with an HPLC purity of 98.0%. Rf=0.1 (petroleum ether/ethyl acetate, V: V=1: 1). 1H NMR (DMSO-d6, 400 MHz) δ: 3.53~3.69 (m, 2H, CH2), 3.87~3.92 (m, 1H, 4'-H), 4.05 (t, J=4.8 Hz, 1H, 3'-H), 4.40~4.45 (m, 1H, 2'-H), 5.26 (t, J=4.8 Hz, 1H, 5'-OH), 5.65 (d, J=8.0 Hz, 1H, 2'-OH), 5.76 (d, J=5.6 Hz, 1H, 1'-H), 6.15 (d, J=5.2 Hz, 1H, ArH), 7.86 (d, J=8.0 Hz, 1H, ArH), 11.34 (s, 1H, ArH); ESI-MS m/z: 269.8 [M+H]+, 291.9 [M+Na]+.
3'-Azido-3'-deoxy-4-deoxy uridine (17): light yellow foam, 78% yield over two steps with an HPLC purity of 95.2%. Rf=0.6 (dichloromethane/methanol, V: V=10: 1). 1H NMR (DMSO-d6, 400 MHz) δ: 3.59~3.66 (m, 1H, CH), 3.80~3.92 (m, 2H, CH, 4'-H), 4.08~4.13 (m, 1H, 3'-H), 4.38~4.43 (m, 1H, 2'-H), 5.39 (t, J=0.8 Hz, 1H, 5'-OH), 5.75 (d, J=2.4 Hz, 1H, 2'-OH), 6.36 (d, J=5.2 Hz, 1H, 1'-H), 6.48~6.52 (m, 1H, ArH), 8.52~8.58 (m, 1H, ArH); ESI-MS m/z: 254.0 [M+H]+, 275.9 [M+Na]+.
1-(3-Azido-3-deoxy-β-D-ribofuranosyl)-5-nitropyridine-2(1H)-one (18): white solid, 65% yield over two steps with an HPLC purity of 95.7%. Rf=0.5 (dichloromethane/ methanol, V: V=10: 1). m.p. 150~155 ℃ (dec.). 1H NMR (DMSO-d6, 400 MHz) δ: 3.62~3.70 (m, 1H, CH), 3.87~4.00 (m, 2H, CH, 4'-H), 4.14~4.21 (m, 1H, 3'-H), 4.37~4.43 (m, 1H, 2'-H), 5.61 (t, J=4.4 Hz, 1H, 5'-OH), 5.90 (d, J=1.6 Hz, 1H, 2'-OH), 6.43 (d, J=5.2 Hz, 1H, 1'-H), 6.51 (d, J=10.0 Hz, 1H, ArH), 8.15 (dd, J1=2.8 Hz, J2=10.2 Hz, 1H, ArH), 9.63 (d, J=2.8 Hz, 1H, ArH); ESI-MS m/z: 319.9 [M+Na]+. HRMS calcd for C10H11-N5NaO6 [M+Na]+ 320.0607, found 320.0605.
3'-Azido-3'-deoxy-2-thiouridine (19): white solid, 72% yield over two steps with an HPLC purity of 98.4%. Rf=0.4 (dichloromethane/methanol, V: V=10: 1). m.p. 155~160 ℃ (dec.). UV-vis (MeOH) λmax: 273 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 3.50~3.70 (m, 2H, CH2), 3.73~3.80 (m, 2H, 4', 3'-H), 4.45~4.48 (m, 1H, 2'-H), 5.44 (t, J=5.2 Hz, 1H, 5'-OH), 5.99 (d, J=8.4 Hz, 1H, ArH), 6.23 (d, J=5.6 Hz, 1H, 2'-OH), 6.54 (d, J=3.2 Hz, 1H, 1'-H), 8.15 (d, J=8.2 Hz, 1H, ArH), 12.65 (s, 1H, NH); ESI-MS m/z: 285.8 [M]+. HRMS calcd for C9H11N5NaO4S [M+ Na]+ 308.0429, found 308.0429.
4.2.9 Synthesis of 6-chloro-9-(2-O-acetyl-5-O-p-toluoyl-3-azido-3-deoxy-β-D-ribofuranosyl)-9H-purine (39)
To a precooled (0 ℃) mixture of 1, 2-di-O-acetyl-5-O-(4-methylbenzoyl)riboside (32, 7.5 g, 19.9 mmol, 1.0 equiv.) and 6-chloropurine (3.38 g, 21.9 mmol, 1.1 equiv.) in anhydrous acetonitrile (100 mL) was added dropwise a solution of 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU) (9.0 mL, 9.13 g, 60.0 mmol, 3.0 equiv.) in anhydrous acetonitrile (30 mL), followed by addition of trimethylsilyl triflate (14.5 mL, 17.8 g, 80.0 mmol, 4.0 equiv.). The reaction mixture was then stirred at 60 ℃ for 4 h. Upon completion of the reaction as monitored by TLC, the reaction mixture was poured into ice water and was treated with saturated sodium bicarbonate and ethyl acetate. The organic phase was separated, and the aqueous phase was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate. The drying agent was filtered off, and the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column using petroleum ether-ethyl acetate (V: V, 5: 1 to 3: 1) as eluents providing product 39 (8.4 g, 89%) as a white solid. ESI-MS m/z: 272.1 [M]+.
4.2.10 Synthesis of 3'-azido-3'-deoxyadenosine (20)
6-Chloro-9-(2-O-acetyl-5-O-p-toluoyl-3-azido-3-deoxy-β-D-ribofuranosyl)-9H-purine (39) (3.0 g, 6.4 mmol) was dissolved in methanol (30 mL) and the solution was saturated with dry ammonia gas at 0 ℃. The reaction mixture was stirred for overnight at 80 ℃, and was concentrated under reduced pressure. The resulting residue was purified by flash chromatography on a silica gel column to afford 1.3 g of compound 20 as a white solid in 70% yield with an HPLC purity of 96.7%. Rf=0.20 (dichloromethane/ methanol, V: V=10: 1). m.p. 200~205 ℃ (Lit.[21] m.p. 208~212 ℃(dec.)). UV-Vis (MeOH) λmax: 260 nm; ESI-MS m/z: 292.9 [M+H]+.
4.2.11 Synthesis of 3'-azido-3'-deoxy-N6-methyl-adeno-sine (21)
6-Chloro-9-(2-O-acetyl-5-O-p-toluoyl-3-azido-3-deoxy-β-D-ribofuranosyl)-9H-purine (39, 4.6 g, 9.7 mmol) was dissolved in methylamine (60 mL) solution in methanol. The reaction mixture was stirred at 80 ℃ overnight. Upon completion of the reaction as monitored by TLC, the reaction solvent was evaporated under reduced pressure. The resulting residue was purified by flash chromatography on a silica gel column to afford 2.4 g of compound 21[22] as a white solid in 82% yield with an HPLC purity of 98.5%. Rf=0.6 (dichloromethane/methanol, V: V=10: 1). m.p. 165~170 ℃ (dec.); UV-vis (MeOH) λmax: 261 nm; ESI-MS m/z: 307.1 [M+H]+, 329.1 [M+Na]+.
4.2.12 Synthesis of 3'-azido-3'-deoxy-N6, N6-dimethyl-adenosine (22)
6-Chloro-9-(2-O-acetyl-5-O-p-toluoyl-3-azido-3-deoxy-β-D-ribofuranosyl)-9H-purine (39, 4.2 g, 8.9 mmol) was dissolved in dimethylamine (20 mL) solution in methanol. The reaction mixture was heated at 100 ℃ overnight. Upon completion of the reaction as monitored by TLC, the reaction solvent was evaporated under reduced pressure. The resulting residue was purified by flash chromatography on a silica gel column to afford 2.4 g of compound 22 as a white solid in 86% yield with an HPLC purity of 98.5%. Rf=0.7 (dichloromethane/methanol, V: V=10: 1). m.p. 185~190 ℃(dec.) (Lit.[12] m.p. 199~201 ℃).
4.2.13 Synthesis of 6-methylpurine-β-D-(3-azido-3-de-oxy)riboside (23)
This compound was synthesized from the glycosylation of 6-methylpurine (1.4 g, 8.8 mmoL, 1.1 equiv.) with 32 (3.0 g, 8.0 mmoL) and the resulted compound was further deprotected resulted in 1.6 g of final product 23 as a white solid in 72% total yield with an HPLC purity of 99.2%. Rf=0.6 (dichloromethane/methanol, V: V=10: 1). m.p. 170~173 ℃(dec.); 1H NMR (DMSO-d6, 400 MHz) δ: 2.73 (s, 3H, CH3), 3.56~3.65 (m, 1H, CH), 3.69~3.76 (m, 1H, CH), 4.00~4.05 (m, 1H, 4'-H), 4.31~4.36 (m, 1H, 3'-H), 4.99~5.05 (m, 1H, 2'-H), 5.27 (t, J=5.2 Hz, 1H, 5'-OH), 6.03 (d, J=5.6 Hz, 1H, 2'-OH), 6.29 (d, J=5.6 Hz, 1H, 1'-H), 8.75 (s, 1H, ArH), 8.80 (s, 1H, ArH); ESI-MS m/z: 292.1 [M+H]+, 314.1 [M+Na]+. HRMS calcd for C11H13N7NaO3 [M+Na]+ 314.0978, found 314.0979.
4.2.14 Synthesis of 3'-amino-3'-deoxyadenosine (24)
This compound was synthesized from compound 20 (2.0 g, 6.8 moL) resulting in 1.6 g of compound 24 as a gray solid in 87% yield with an HPLC purity of 96.6%. Rf=0.60 (dichloromethane/methanol, V: V=2: 1). m.p. 230~235 ℃(dec.) (Lit.[21] > 230 ℃); UV-Vis (MeOH) λmax: 258 nm; ESI-MS m/z: 267.1 [M+H]+, 329.1 [M+Na]+.
4.2.15 Synthesis of 1, 2-di-O-acetyl-5-O-(4-methyl benzoyl)-L-riboside (43)
1, 2-O-Isopropylidene-α-L-xylofuranose (40) was synthesized from L-(-)-xylose by two steps in 98% total yield according to procedures described in the literature.[15] This material (100 g, 0.53 mol) was dissolved in 800 mL of dry pyridine and the mixture was cooled to 0 ℃. Then 4-methylbenzoyl chloride (76.3 mL, 0.58 mol, 1.1 equiv.) was added dropwise with stirring. The reaction mixture was stirred for 2 h. Upon completion of the reaction as monitored by TLC, the reaction mixture was poured into ice water and was treated with ethyl acetate. The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column to provide product 1, 2-O-isopropylidene-5-O-(4-methylbenzoyl)-α-L-xylofura-nose as a colorless oil. It was then dissolved in 200 mL of dry pyridine and 600 mL of dichloromethane. The mixture was cooled to -20 ℃. Then trifluoromethanesulfonic anhydride (96.5 mL, 0.58 mol, 1.1 equiv.) was added dropwise with stirring. The reaction mixture was stirred for 2 h at -20 ℃. Upon completion of the reaction as monitored by TLC, the reaction mixture was poured into saturated sodium bicarbonate and was treated with ethyl acetate. The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column to provide 178 g of product 1, 2-O-isopropylidene-5-O-(4-methylbenzoyl)-3-O-trifluoromethanesulfonyl-α-L-xylo-furanose (41) in 76.5% yield in two steps as a white solid. This material was dissolved in 1500 mL of dry N, N-dimethylformamide (DMF), and sodium azide (52.0 g, 0.81 moL, 2.0 equiv.) was added. The reaction mixture was stirred for 72 h at room temperature. The reaction mixture was poured into water and was treated with ethyl acetate. The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column to provide 74.0 g of product 3-azido-1, 2-O-isopropylidene-5-O-(4-methylbenzoyl)-3-eoxy-α-L-xylofuranose (42) in 55% yield as a colorless oil. This material was dissolved in 200 mL of acetic acid and 100 mL of acetic anhydride, and 3.0 mL of concentrated sulfuric acid was added under stirring. The reaction mixture was stirred at room temperature overnight, and treated with 200 mL water. It was neutralized with sodium bicarbonate carefully and extracted with dichloromethane. The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by flash chromatography on a silica gel column using petroleum ether-ethyl acetate as an eluent to give 63.0 g white sticky product 43 in 75% yield.
4.2.16 Synthesis of 3'-azido-3'-deoxy-L-nucleoside derivatives 44~46 and deprotected compounds 25~27
These compounds were synthesized by the glycosylation of uracil, 5-methyl uracil and 4-fluorouracil with 43, respectively, resulting in compounds 44~46. Further deprotection resulted in final products 25~27 as white solids in 65%~72% overall yield.
3'-Azido-3'-deoxy-β-L-uridine (25): white solid, 73% yield in two steps with an HPLC purity of 96.8%. Rf=0.5 (dichloromethane/methanol, V: V=10: 1). m.p. 135~138 ℃; UV-Vis (MeOH) λmax: 259 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 3.49~3.54 (m, 2H, CH2), 3.80~4.05 (m, 2H, 4', 3'-H), 4.35~4.50 (m, 1H, 2'-H), 5.29 (t, J=5.2 Hz, 1H, 5'-OH), 5.66 (d, J=8.0 Hz, 1H, ArH), 5.76 (d, J=5.2 Hz, 1H, 2'-OH), 6.17 (d, J=5.2 Hz, 1H, 1'-H), 7.88 (d, J=8.0 Hz, 1H, ArH), 11.48 (s, 1H, NH); ESI-MS m/z: 270.2 [M+H]+, 291.9 [M+Na]+.
3'-Azido-3'-deoxy-5-methyl-β-L-uridine (26): white foam, 77% yield in two steps with an HPLC purity of 98.9%. Rf=0.6 (dichloromethane/methanol, V: V=10: 1). UV-Vis (MeOH) λmax: 260 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 1.77 (s, 3H, CH3), 3.53~3.71 (m, 2H, CH2), 3.85~3.89 (m, 1H, 4'-H), 4.07 (t, J=5.2 Hz, 1H, 3'-H), 4.40~4.45 (m, 1H, 2'-H), 5.26 (t, J=5.2 Hz, 1H, 5'-OH), 5.76 (d, J=5.6 Hz, 1H, 2'-OH), 6.11 (d, J=5.6 Hz, 1H, 1'-H), 7.71 (s, 1H, ArH), 11.34 (s, 1H, NH); ESI-MS m/z: 284.1 [M+H]+, 306.1 [M+Na]+.
3'-Azido-3'-deoxy-5-fluoro-β-L-uridine (27): white solid, 70% yield in two steps with an HPLC purity of 97.5%. Rf=0.6 (dichloromethane/methanol, V: V=10: 1). m.p. 160~163 ℃; UV-Vis (MeOH) λmax: 261 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 3.55~3.64 (m, 1H, CH), 3.68~3.76 (m, 1H, CH), 3.90~3.98 (m, 1H, 4'-H), 4.03 (t, J=5.2 Hz, 1H, 3'-H), 4.37~4.52 (m, 1H, 2'-H), 5.44 (t, J=4.8 Hz, 1H, 5'-OH), 5.69~5.73 (m, 1H, 2'-OH), 6.19 (d, J=5.6 Hz, 1H, 1'-H), 8.27 (d, J=7.2 Hz, 1H, ArH), 11.89 (s, 1H, NH); ESI-MS m/z: 288.2 [M+H]+, 310.1 [M+Na]+; HRMS calcd for C9H10FN5NaO5 [M+Na]+310.0564, found 310.0563.
4.2.17 General procedure for the synthesis of 3'-azido-3'-deoxy-L-cytidine derivatives 28~30
Compounds 28~30 were synthesized from compounds 44~46, respectively.
3'-Azido-3'-deoxy-β-L-cytidine (28): white solid, 78% yield in two steps with an HPLC purity of 98.4%. Rf=0.3 (dichloromethane/methanol, V: V=5: 1). m.p. 140~143 ℃(dec.); UV-vis (MeOH) λmax: 265 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 3.53~3.60 (m, 1H, CH), 3.65~3.71 (m, 1H, CH), 3.88~3.97 (m, 2H, 3'-H, 4'-H), 4.33~4.39 (m, 1H, 2'-H), 5.21 (t, J=5.2 Hz, 1H, 5'-OH), 5.70~5.77 (m, 2H, 2'-OH, ArH), 6.10 (d, J=5.2 Hz, 1H, 1'-H), 7.20 (s, 1H, NH), 7.30 (s, 1H, NH), 7.82 (d, 1H, J=7.6 Hz, ArH); ESI-MS m/z: 269.1 [M+H]+.
3'-Azido-3'-deoxy-5-methyl-β-L-cytidine (29): light yellow solid, 74% yield in two steps with an HPLC purity of 95.3%. Rf=0.45 (dichloromethane/methanol, V: V=5: 1). m.p. 105~107 ℃; UV-vis (MeOH) λmax: 275 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 1.87 (s, 3H, CH3), 3.54~3.62 (m, 1H, CH), 3.68~3.77 (m, 1H, CH), 3.90~4.00 (m, 2H, 4', 3'-H), 4.37~4.40 (m, 1H, 2'-H), 5.31 (t, J=5.2 Hz, 1H, 5'-OH), 5.75 (d, J=4.4 Hz, 1H, 2'-OH), 6.14 (d, J=5.2 Hz, 1H, 1'-H), 7.72 (s, 1H, NH), 7.81 (s, 1H, NH), 7.84 (s, 1H, ArH); ESI-MS m/z: 283.1 [M+H]+.
3'-Azido-3'-deoxy-5-fluoro-β-L-cytidine (30): white foam, 74% yield in two steps with an HPLC purity of 97.3%. Rf=0.5 (dichloromethane/methanol, V: V=5: 1). UV-vis (MeOH) λmax: 239, 280 nm; 1H NMR (DMSO-d6, 400 MHz) δ: 3.54~3.62 (m, 1H, CH), 3.70~3.76 (m, 1H, CH), 3.90~3.98 (m, 2H, 3'-H, 4'-H), 4.31~4.37 (m, 1H, 2'-H), 5.38 (br, 1H, 5'-OH), 5.70~5.74 (m, 1H, 2'-OH), 6.15 (br, 1H, 1'-H), 7.61 (s, 1H, NH), 7.81 (s, 1H, NH), 8.17 (d, 1H, J=7.2 Hz, ArH); ESI-MS m/z: 287.1 [M+H]+, 573.2 [2M+H]+.
4.2.18 Synthesis of 6-chloro-9-[2-O-acetyl-5-O-(p-toluoyl)-3-azido-3-deoxy-β-L-ribofuranosyl]-9H-purine (47) and 3'-azido-3'-deoxy-β-L-adenosine (31)
Compound 47 was synthesized from 6-chloropurine (1.80 g, 11.7 mmoL, 1.1 equiv.) and 43 (4.0 g, 10.6 mmoL, 1.0 equiv.). Further deprotection by Method 6 resulted in 2.2 g of final product 31 as a white solid in 70% over yield with an HPLC purity of 97.0%. Rf=0.4 (dichloromethane/methanol, V: V=10: 1). m.p.[21] 191~195 ℃(dec.); UV-vis (MeOH) λmax: 259 nm; ESI-MS m/z: 293.1 [M+H]+, 315.2 [M+H]+.
Acknowledgements The authors thank Sherry Li, Zhuo Li, Kewen Zhu, Hailong Du, Ningjie Chang for their effort.
Supporting Information 1H NMR, MS and HRMS spectral data. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
-
-
[1]
(a) Silvestris, N. ; Daprile, M. ; Andreola, G. ; Locopo, N. ; Marini, L. ; Cructitta, E. ; Delena, M. ; Lorusso, V. Int. J. Oncol. 2004, 24, 389.
(b) Riggs, A. D. ; Jones, P. A. Adv. Cancer Res. 1983, 40, 1.
(c) Momparler, R. L. ; Momparler, L. F. Cancer Chemother. Pharmacol. 1989, 25, 51.
(d) Asselah, T. Expert Opin. Pharmacother. 2014, 15, 121. -
[2]
(a) Mitsuya, H. ; Weinhold, K. J. ; Furman, P. A. ; St Clair, M. H. ; Lehrman, S. N. ; Gallo, R. C. ; Bolognesi, D. ; Barry, D. W. ; Broder, S. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 7096.
(b) De Clercq, E. Biochem. Pharmacol. 1994, 47, 155. -
[3]
(a) Wang, Q. ; Hu, W. -D. ; Wang, S. -Y. ; Pan, Z. -L. ; Tao, L. ; Guo, X. -H. ; Qian, K-D. ; Chen, C. -H. ; Lee, K-H. ; Chang, J. -B. Eur. J. Med. Chem. 2011, 46, 4178.
(b) Zheng, L. -Y. ; Wang, Q. ; Yang, X. -R. ; Guo, X. -H. ; Chen, L. ; Tao, L. ; Dong, L. -H. ; Li, Y. -J. ; An, H. ; Yu, X. -J. ; Wang, Q. -D. ; Chang, J. -B. Antiviral Ther. 2012, 17, 679. -
[4]
(a) Klinchan, C. ; Hsu, Y. -L. ; Lo, L-C. ; Pluempanupat, W. ; Chuawong, P. Tetrahedron Lett. 2014, 55, 6207.
(b) Schirmeister-Tichya, H. ; Iaconoc, K. T. ; Mutoc, N. F. ; Homanb, J. W. ; Suhadolnikb, R. J. ; Pfleiderer, W. Helv. Chim Acta 1999, 82, 597.
(c) Zhang, L. ; Cui, Z. -Y. ; Zhang, B. -L. Helv. Chim. Acta 2003, 86, 703.
(d) Robins, M. J. ; Hawrelak, S. D. ; Hernández, A. E. ; Wnuk, S. F. Nucleosides, Nucleotides Nucleic Acids 1992, 11, 821. -
[5]
(a) Zatsepin, T. S. ; Stetsenko, D. A. ; Gait, M. J. ; Oretskaya, T. S. Bioconjugate Chem. 2005, 16, 471.
(b) Matulic-Adamic, J. ; Serebryany, V. ; Haeberli, P. ; Mokler, V. R. ; Beigelman, L. Bioconjugate Chem. 2002, 13, 1071. -
[6]
(a) Roy, V. ; Zerrouki, R. ; Krausz, P. ; Laumond, G. ; Aubertin, A. M. Nucleosides, Nucleotides Nucleic Acids 2007, 26, 413.
(b) Soenens, J. ; Fran ois, G. F. ; Eeckhout, E. V. ; Herdewijn, P. Nucleosides, Nucleotides Nucleic Acids 1995, 14, 409.
(c) Ozols, A. M. ; Azhayev, A. V. ; Dyatkina, N. B. ; Krayevsky, A. A. Synthesis 1980, 557.
(d) Liang, C. W. ; Kim, M. J. ; Jeong, L. S. ; Chun, M. W. Nucleosides, Nucleotides Nucleic Acids 2003, 22, 2039; -
[7]
Arya, A.; Mathur, D.; Tyagi, A.; Kumar, R.; Kumar, V.; Olsen, C. E.; Saxena, R. K.; Prasad, A. K. Nucleosides, Nucleotides Nucleic Acids 2013, 32, 646. doi: 10.1080/15257770.2013.847957
-
[8]
(a) Lin, T. -S. ; Yang, J. -H. ; Liu, M. -C. ; Shen, Z. -Y. ; Cheng, Y. -C. ; Prusoff, W. H. ; Birnbaum, G. I. ; Giziewicz, J. ; Ghazzouli, I. ; Brankovan, V. ; Feng, J. -S. ; Hsiun, G. -D. J. Med. Chem. 1991, 34, 693.
(b) Greco, N. J. ; Tor, Y. Tetrahedron 2007, 63, 3515. -
[9]
(a) Handschumacher, R. E. J. Bid. Chem. 1960, 235, 764.
(b) Capek, A. ; Svatek, E. ; Tadra, M. Folia Microbiol. 1963, 8, 304.
(c) Skoda, J. Prog. Nucleic Acid Res. Mol. Biol. 1963, 2, 197. -
[10]
(a) Robins, M. L. ; Currie, B. L. ; Robins, R. K. ; Bloch, A. Proc. Am. Assoc. Cancer Res. 1969, 10, 73.
(b) McPartland, R. P. ; Wang, M. C. ; Bloch, A. ; Weinfeld, H. Cancer Res. 1974, 34, 3107.
(c) Khare, G. P. ; Sidwell, R. W. ; Huffman, J. H. ; Tolman, R. L. ; Robins, R. K. Proc. Soc. Exp. Biol. Med. 1972, 140, 880.
(d) Shannon, W. M. ; Brockman, R. W. ; Westbrook, L. ; Shaddix, S. ; Schabel, F. M. Jr. J. Natl. Cancer Inst. 1974, 52, 199. -
[11]
(a) Zhou, L. ; Cheng, X. ; Connolly, B. A. ; Dickman, M. J. ; Hurd, P. J. ; Hornby, D. P. J. Mol. Biol. 2002, 321, 591.
(b) Driscoll, J. S. ; Marquez V. E. ; Plowman, J. ; Liu, P. S. ; Kelley, J. A. ; Barchi, J. J. Jr. J. Med. Chem. 1991, 34, 3280. -
[12]
Robins, M. J.; Miles, R. W.; Samano, M. C.; Kaspar, R. L. J. Org. Chem. 2001, 66, 8204. doi: 10.1021/jo010935d
-
[13]
Kim, B.-T.; Kim, S.-K.; Lee, S.-J.; Hwang, K.-J. Bull. Korea Chem. Soc. 2004, 25, 243; doi: 10.5012/bkcs.2004.25.2.243
-
[14]
(a) Chong, Y. ; Gumina, G. ; Mathew, J. S. ; Schinazi, R. F. ; Chu, C. K. J. Med. Chem. 2003, 46, 3245.
(b) Ma, T. -W. ; Pai, S. B. ; Zhu, Y. L. ; Lin, J. S. ; Shanmuganathan, K. ; Du, J. -F. ; Wang, C. -G. ; Kim, H. -B. ; Newton, M. G. ; Cheng, Y. C. ; Chu, D. K. J. Med. Chem. 1996, 39, 2835.
(c) Kotra, L. P. ; Xiang, Y. -J. ; Newton, M. G. ; Schinazi, R. F. ; Cheng, Y. -C. ; Chu, C. K. J. Med. Chem. 1997, 40, 3635. -
[15]
Cooperwood, J. S.; Boyd, V.; Gumina, G.; Chu, C. K. Nucleosides, Nucleotides Nucleic Acids 2000, 19, 219. doi: 10.1080/15257770008033005
-
[16]
(a) Hassan, A. E. A. ; Abou-Elkair, R. A. I. ; Montgomery, J. A. ; Secrist Ⅲ, J. A. Nucleosides, Nucleotides Nucleic Acids 2000, 19, 1123.
(b) Marasco, C. J. Jr. ; Pera, P. J. ; Spiess, A. J. ; Bernacki, R. ; Sufrin, J. R. Molecules 2005, 10, 1015. -
[17]
Rompaey, P. V.; Nauwelaerts, K.; Vanheusden, V.; Rozenski, J.; Munier-Lehmann, H.; Herdewijn, P.; Calenbergh, S. V. Eur. J. Org. Chem. 2003, 2911. doi: 10.1002/ejoc.200300177/full
-
[18]
Ora, M.; Mattila, K.; Lonnberg, T.; Oivanen, M.; Lonnberg, H. J. Am. Chem. Soc. 2002, 124, 14364. doi: 10.1021/ja027499c
-
[19]
Urata, Y.; Mitsu, Y.; Nakamura, K. T. Chem. Pharm. Bull 1981, 29, 2769. doi: 10.1248/cpb.29.2769
-
[20]
Gadthula, S.; Chu, C. K.; Schinazi, R. F. Nucleosides, Nucleotides, Nucleic Acids 2005, 24, 1707. doi: 10.1080/15257770500267170
-
[21]
Botta, O.; Moyroud, E.; Lobato, C.; Strazewski, P. Tetrahedron 1998, 54, 13529. doi: 10.1016/S0040-4020(98)00819-9
-
[22]
Gao, Z.-G.; Duong, H. T.; Sonina, T.; Kim, S.-K.; Rompaey, P. V.; Calenbergh, S. V.; Mamedova, L.; Kim, H. O.; Kim, M. J.; Kim, A. Y.; Liang, B. R.; Jeong, L. S.; Jacobson, K. A. J. Med. Chem. 2006, 49, 2689. doi: 10.1021/jm050968b
-
[1]
-

Scheme 1 Synthesis of 3'-azido-3'-deoxy-D-pyrimidine nucleosides 1~14
Reagents and conditions: (a) Base, BSA, CH3CN, r.t.; TMSOTf, r.t.; (b) NH3/MeOH, r.t.; (c) Pd/C, H2; (d) POCl3, 1, 2, 4-triazole, Et3N, r.t.; (e) NH3/MeOH, r.t.; (f) ICl, NaN3, CH3CN; (g) TsCl, pyridine; (h) DBU, CH3CN, reflux; (i) NH4OH, r.t.

Scheme 2 Synthesis of 3'-azido-3'-deoxy-D-nucleosides 15~19 with special heterocyclic bases
Reagents and conditions: (a) Base, BSA, CH3CN, r.t.; TMSOTf, r.t.; (b) NH3/ MeOH, r.t.

Scheme 3 Synthesis of 3'-azido-3'-deoxy-D-purine nucleosides 20~24
Reagents and conditions: (b) NH3/MeOH, r.t.; (c) Pd/C, H2; (j) Base, DBU, TMSOTf, r.t.~60 ℃; (k) NH3/MeOH, 80 ℃ (to 20); (l) NH2Me/ MeOH, 80 ℃ (to 21); (m) NHMe2/MeOH, 100 ℃ (to 22)
-


 下载:
下载:




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 12
- 文章访问数: 2758
- HTML全文浏览量: 200
 下载:
下载:
