 图式1
经典的Ugi四组分反应
Scheme1.
Classical Ugi four-component reaction
图式1
经典的Ugi四组分反应
Scheme1.
Classical Ugi four-component reaction
引用本文:
李修明, 贾学顺, 殷亮. Post-Ugi反应的研究进展[J]. 有机化学,
2017, 37(9): 2237-2249.
doi:
10.6023/cjoc201704026

Citation: Li Xiuming, Jia Xueshun, Yin Liang. Recent Progress on Post-Ugi Reaction[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2237-2249. doi: 10.6023/cjoc201704026
Citation: Li Xiuming, Jia Xueshun, Yin Liang. Recent Progress on Post-Ugi Reaction[J]. Chinese Journal of Organic Chemistry, 2017, 37(9): 2237-2249. doi: 10.6023/cjoc201704026
Post-Ugi反应的研究进展
摘要:
Ugi反应是一种高效的、原子经济性的多组分反应.Post-Ugi反应作为其拓展,是多取代杂环化合物的有效合成方法,受到人们的广泛关注.综述了post-Ugi反应的发展,重点总结了近些年来的相关报道.
-
关键词:
- post-Ugi反应
- / 杂环
- / 加成
- / 偶联
English
Recent Progress on Post-Ugi Reaction
Abstract:
Ugi reaction is an effective and atom-economical multicomponent reaction. The sequences of Ugi multicomponent reactions and following various postcondensation transformations constitute an extremely powerful synthetic method for heterocyclic compounds with elaborate substitution patterns. Herein, the development in this field is summarized.
-
Key words:
- post-Ugi reaction
- / heterocycle
- / addition
- / coupling
-
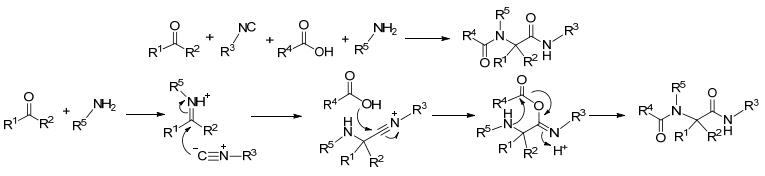
根据起始原料数量的不同, 有机化学反应可以分为:单组分反应、两组分反应、多组分反应和聚合反应.单组分反应只涉及一种起始原料(催化剂除外), 反应生成一种或两种产物. Claisen重排就是一种典型的单组分反应.在两组分反应中, 两种起始原料反应得到一种产物.多组分反应(Multicomponent reaction, MCR)通常定义为三种或者三种以上的反应物以一锅法发生化学反应, 并且生成的产物中含有所有起始原料片段的一种合成方法[1]. MCR条件温和、易于操作, 一般不需要无水、无氧等严格的反应条件.与传统的多步合成方法不同, MCR能够在一个操作过程中产生多于两个的化学键, 实现从简单的原料出发快速地构建结构具有多样性和复杂性的化合物. MCR还是一种建立小分子化合物库的强有力工具, 它是构效关系研究不可或缺的一部分. MCR能够产生一些不常见的化合物, 将有助于生物和医药领域的研究[2~6].
1959年, 德国化学家Ugi等[7]首次报道了醛(酮)、胺、羧酸、异腈间的一锅法反应即Ugi四组分反应(Ugi four-component reaction, U-4CR) (Scheme 1).反应通常以较高的反应浓度在甲醇或三氟乙醇中进行.机理是:醛(酮)和胺脱水生成亚胺, 异腈对亚胺亲核进攻形成腈鎓离子中间体, 然后它再和羧酸反应, 最后通过分子内的Mumm重排得到双酰胺产物(Scheme 1).此反应条件温和、操作简便、收率较高, 被广泛应于多肽和其它小分子库的固相及液相合成中, 成为有机合成化学中一个重要的反应[8~10].
Ugi反应原料的多样性导致了其产物的多样性.根据R基团的不同, 将Ugi反应与不同的反应串联, 可扩大反应的应用性.这种串联即为post-Ugi反应, 是多取代杂环和多肽的有效合成方法, 受到人们的广泛关注.近年来, 已有综述性论文对各种基于异腈的多组分反应进行了专门的评述[11~15].最近, 郭长彬课题组[16]还对Ugi反应脱Boc保护基环合策略在含氮杂环合成方面进行了综述.本文将重点围绕post-Ugi反应, 对其最近几年的发展做简要介绍.
图式1
经典的Ugi四组分反应
Scheme1.
Classical Ugi four-component reaction
1 叠氮参与的post-Ugi反应
1.1 Azido-Ugi反应
1961年, 德国化学家Ugi[17]首次用叠氮酸(由TMSN3原位生成)代替经典Ugi反应中的羧酸合成了含四氮唑的化合物, 即azido-Ugi反应.机理是:异腈对醛(或酮)和胺缩合形成的亚胺进攻得到腈鎓离子中间体, 然后叠氮酸代替羧酸对该中间体进攻, 再经过重排, 以一锅法得到四氮唑产物.在报道之初的一段时间, 对这种改良的Ugi反应相关研究比较少[18~20].原因可能是该方法对含四氮唑杂环的合成不是十分高效.含四氮唑的苯并二氮杂环化合物是血小板聚集的有效抑制剂, 对神经肽缩胆囊素也表现出了抑制作用.最近, 一些利用azido-Ugi反应合成此类杂环化合物的策略相继被报道, 例如:哌嗪并四氮唑1[21]、苯并哌嗪四氮唑2[22]以及其它含四氮唑的杂环化合物3[23].
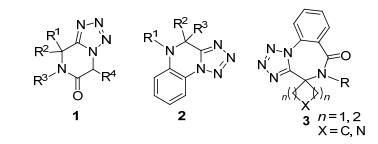 图1
含四氮唑化合物1~3的结构
Figure1.
Structures of compounds 1~3 containing tetrazole
图1
含四氮唑化合物1~3的结构
Figure1.
Structures of compounds 1~3 containing tetrazole
2010年, Voskressensky课题组[24]发展了通过azido-Ugi反应来合成二氮

环亚胺是一种重要的有机合成砌块, 它可以将环胺引入到分子中.这些分子可以进一步构建含有氨基片段的天然产物类似物. 2013年, Nenajdenk等[25]将α全氟烷基的环亚胺应用于azido-Ugi反应, 合成了含全氟烷基的环胺基四氮唑(Eq. 2).环亚胺、TMSN3和异腈在甲醇中于室温下反应即可以以中等的产率生成5-环胺基四氮唑5.与五元或七元的α-CF3环亚胺相比, 六元环亚胺具有更好的反应性, 反应产率最高.当全氟烷基由CF3变为C2F5时, 反应的产率也随之降低.他们认为这是受到了C2F5更大的立体位阻的影响.
2013年, Nenajdenko课题组[26]报道了基于2-取代环亚胺的azido-Ugi反应, 该反应可以用于制备四氮唑取代的五元、六元和七元环胺. 2-取代的环亚胺、异腈和在甲醇中原位生成的HN3, 在室温下反应以中等收率合成了四氮唑的环胺产物(Scheme 2).反应的不足之处是:环亚胺上的R1基团必须是给电子基, 对于R1基团为氢原子或吸电子基的底物, 反应不能进行.他们的机理研究表明:相对于异腈, 亚胺对反应的影响更大.四氮唑上的保护基可以在催化氢化的条件下脱保护, 定量地生成5-环胺基-1H-四氮唑.它克服了脯氨酸在常见溶剂中不溶解的缺点, 是一类重要的有机催化剂[27].另外, 他们还利用酒石酸在乙醇中完成了两种对映体的手性拆分.随后氢化脱苄基化, 完成了一系列5-环胺基-1H-四氮唑的合成.值得一提的是, 他们还将其作为有机小分子催化剂成功地用于偶氮二酸酯与醛进行的不对称胺化反应, 当R=Et时, ee=92%; R=t-Bu时, ee≥99% (Scheme 2).
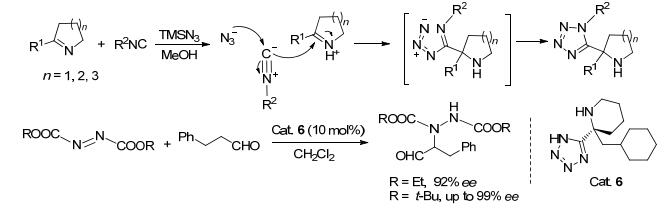 图式2
Azido-Ugi反应合成四氮唑及其作为有机小分子催化剂的应用
Scheme2.
Synthesis of tetrazole-derived heterocycles via azido-Ugi reaction and its application as organocatalyst
图式2
Azido-Ugi反应合成四氮唑及其作为有机小分子催化剂的应用
Scheme2.
Synthesis of tetrazole-derived heterocycles via azido-Ugi reaction and its application as organocatalyst
1963年, Ugi等[28]发现以环己烯基异腈为原料的多组分反应产物, 其C—N键在酸性条件下可以被切断.随之, 他提出了可转化的异腈(convertible isocyanide)的概念, 然后这个概念被不断地发展.一些可以被转化或切断的含有异腈官能团的片段被报道.对于可转化的异腈, 其所得Ugi产物中的酰胺可转化为酯、硫酯、酮和羧酸等基团.对于Ugi产物的C—N键的切断, 存在反应条件剧烈和原子经济性差等缺点[29, 30].
2016年, D mling等[31]首次将β-腈基异腈应用于azido-Ugi反应. Ugi产物在LiOH的作用下以四氢呋喃(THF)/H2O为溶剂反应30 min, 脱去β-氰基乙基, 完成了温和条件下的快速脱保护(Scheme 3).该反应具有良好的收率和较宽的底物范围. β-氰基乙基脱去的机理是:碱攫取腈基的α氢, 然后电子流向四氮唑, 接着C—N键断开生成四氮唑负离子和高挥发性丙烯腈.作者进一步的研究发现, 四氮唑阴离子的共振稳定作用是反应能够进行的关键.这种方法仅适用于含有四氮唑的底物,
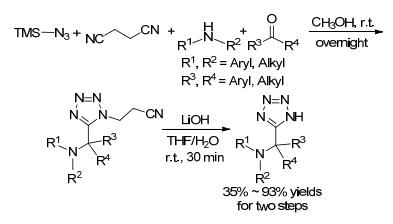 图式3
可切断的β-腈基异腈应用于Ugi反应
Scheme3.
Application of cleavable β-cyanoethyl isocyanide in Ugi reaction
图式3
可切断的β-腈基异腈应用于Ugi反应
Scheme3.
Application of cleavable β-cyanoethyl isocyanide in Ugi reaction
对于更普遍的Ugi反应产物, 反应是不能进行的.另外, 1H-四氮唑是羧酸的生物电子等排体, 在药物设计中有重要的作用.这种策略可以作为现有合成方法有价值的替代.
1.2 Joullié-Ugi反应
Joullié-Ugi三组分反应最早是1982年由Joullié等[32]发展的环亚胺、羧酸和异腈反应合成α位取代的哌啶酰胺或四氢吡咯酰胺的一种方法.反应的产物有两种非对映异构体. Flanagan和Joullié[33]在其开创性的研究中发现, 溶剂的极性对反应的非对映选择性影响较大, 极性溶剂中的产物以位阻较大的cis异构体为主, 而在弱极性溶剂中反之.他们认为这种现象是不同极性的溶剂对羧酸和亚胺形成的“导向离子对”的稳定作用不同所致.
最近, Ichikawa等[34]在Flanagan和Joullié工作的基础上用α位硅氧基的五元环亚胺对该反应的机理进行了详细的研究.他们首先考察了溶剂对反应非对映选择性的影响.他们发现在弱极性的溶剂中产物主要以cis-7为主, 在强极性的醇类溶剂中产物主要以trans-7为主, 在六氟异丙醇中trans/cis产物的比例甚至能>99:1.然后他们考察了羧酸和异腈对反应的影响, 结果表明羧酸的结构对反应的非对映选择性影响不大, 而富电子的异腈在六氟异丙醇中显示了较高的非对映选择性.进一步的对比实验表明:带有供电子基的异腈可以加速反应.随后的Hammit曲线研究表明溶剂不同反应所经的历程也不同.在弱极性溶剂中, 羧酸对亚胺的质子化形成紧密离子对Ⅰ, 然后异腈从离子对中间体β面进攻得到cis产物; 而在强极性溶剂中, 羧酸和亚胺形成溶剂分离的离子对Ⅱ, 异腈从其α面进攻得到trans产物.在弱极性溶剂(如甲苯)中的反应历程是path 1;而在强极性的醇类溶剂(如六氟异丙醇)中的反应历程是path 1和path 2, 但以后者为主(Scheme 4).
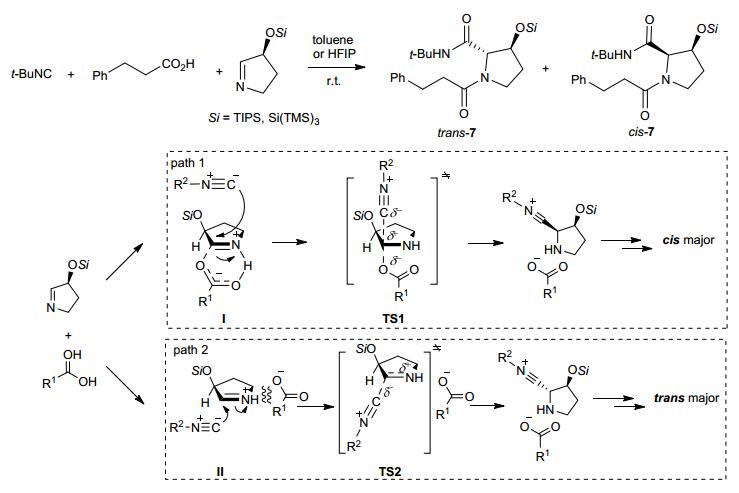 图式4
Joullié-Ugi反应可能的反应历程
Scheme4.
Proposed mechanism of Joullié-Ugi reaction
图式4
Joullié-Ugi反应可能的反应历程
Scheme4.
Proposed mechanism of Joullié-Ugi reaction
2004年, Riva等[35]以单取代的手性环亚胺为原料通过Joullié-Ugi反应合成了2, 5-二取代的吡咯烷(Eq. 3).反应以中等产率生成了两种非对映异构体.该小组尝试用大位阻的R3来控制反应的非对映选择性, 但其dr值仅介于53:47和68:32之间. Joullié-Ugi反应的非对映选择性一般很难由单取代的手性环亚胺通过底物控制[36, 37].二取代的手性环亚胺参与的Ugi反应, 其非对映选择性却可在底物的作用下得到很好地控制[38, 39]. 2010年, Orru等[40]报道了顺式的双环亚胺参与的Joullié-Ugi反应, 其非对映选择性和对映选择性在底物的作用下都得到了很好的控制(Eq. 4).
1.3 Ugi/click反应
有机叠氮化合物和炔基化合物在加热条件下能发生1, 3-偶极环加成来构建不饱和五元三氮唑即Huisgen反应.传统的Huisgen反应有很多缺点, 例如反应时间长、反应温度高、选择性差等.后来Sharpless改进了这一反应, 在室温下即可以较高的产率高选择性地生成1, 4-二取代1, 2, 3-三氮唑.在此类反应的基础上, 他提出了点击化学(Click chemistry)的概念, 指易于操作、产率高、选择性好的化学反应.改进后的一价铜催化的末端炔和有机叠氮的Huisgen环加成反应, 可选择性地生成1, 2, 3-二取代的三氮唑, 具有反应条件温和、产率高等优点, 被认为是click反应中的精华, 现已在药物开发、新材料的合成中得到广泛的应用.一价铜催化的末端炔和叠氮化物的Huisgen环加成被用来特指click反应[41]. Ugi反应和click反应的结合已经引起了许多人的关注.
同时含有异腈和叠氮基的化合物, 一方面可以通过异腈转化为多肽, 另一方面还可以通过叠氮基参与其它反应. 2010年, Nenajdenko等[42]报道了将由L-氨基醇制得的叠氮基异腈引入到了Ugi/click反应中.第一步, 叠氮基异腈和脂肪醛、芳基羧酸、胺在甲醇中于室温反应12 h以较高的产率和高的非对映选择性(>99% de)得到相应的Ugi产物; 第二步, 在10%的CuI•P(OEt)3催化下, 于60 ℃反应3 h得到环加成产物(Scheme 5).尽管叠氮异腈是高含氮化合物, 但其在室温下是比较稳定的.另外, 叠氮异腈结构本身是一种良好的双齿配体, 能够和铜催化剂形成稳定的络合物从而使催化剂失活, 因而不能直接用于铜催化的反应. 2012年, 该小组[43]还报道了一种通过叠氮基肽和含CF3炔的1, 3-偶极环加成来合成含α-CF3的四肽的方法, 成功地将三氟甲基引入到了多肽类似物中, 为制备含三氟甲基的化合物提供了新方法.
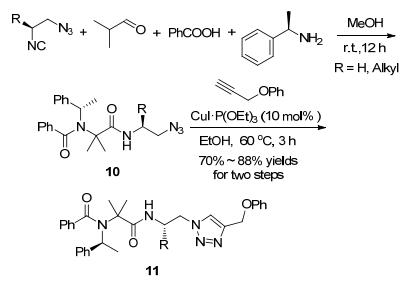 图式5
Nenajdenko报道的Ugi/click反应序列
Scheme5.
The Ugi/click sequence reported by Nenajdenko
图式5
Nenajdenko报道的Ugi/click反应序列
Scheme5.
The Ugi/click sequence reported by Nenajdenko
随后, Pramitha课题组[44]报道了在Ugi产物中引入叠氮基, 然后进行环加成的反应序列(Scheme 6).作者首先将氯代的醋酸、叔丁基异腈、醛和胺反应得到氯代多肽12, 然后在二氯甲烷中和叠氮化钠在碱性条件下反应生成叠氮基取代的产物13.最后, 13和14在由抗坏血酸钠还原硫酸铜原位生成的一价铜的催化下反应生成了环加成产物15.反应前两步收率均在中等或中等以上, 最后一步接近于定量.
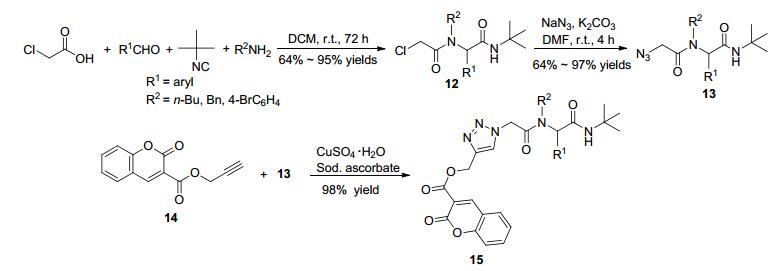 图式6
Pramitha报道的Ugi/click反应序列
Scheme6.
The Ugi/click sequence reported by Pramitha
图式6
Pramitha报道的Ugi/click反应序列
Scheme6.
The Ugi/click sequence reported by Pramitha
2012年, 蔡春课题组[45]将二乙烯酮加入到Ugi/click连续反应体系中, 合成了5-甲基-1H-1, 2, 3-三唑修饰的肽类似物, 实现了不需要铜催化剂的环加成反应(Scheme 7). Ugi反应之后, 直接在反应体系中加入二乙烯酮、碱和伯胺, 经过回流反应可得到环加成的产物17.两步的总产率在31%到61%之间, 反应底物适用性广.药物合成中残留的铜催化剂往往对细菌和哺乳动物细胞是有毒的.这种不需要铜催化剂的策略将有助于药物发现和药物合成.最后, 作者还用Lipinski法对由该反应得到的一系列化合物的类药性进行了评价.结果表明大部分的化合物符合Lipinski五规则.
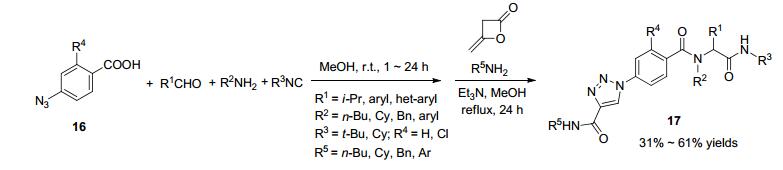 图式7
一锅法合成三唑修饰的肽类似物
Scheme7.
Synthesis of triazole-modified peptidomimetics through one-pot strategy
图式7
一锅法合成三唑修饰的肽类似物
Scheme7.
Synthesis of triazole-modified peptidomimetics through one-pot strategy
环状肽类似物对蛋白酶的降解具有抗性, 并显示出高度的构象稳定性.此类多肽的构象在空间上受到环的限制, 减少了其药效基团和作用位点形成复合物的熵损失, 增强了二者的亲和力.因此, 对这类化合物的开发引起了人们相当大的兴趣[46].从非环分子获得大环的最常见方法是内酯化和内酰胺化, 其它策略还包括烯烃复分解(RCM)、环加成等, 其中环加成以click反应为主.分子内Ugi反应和click反应串联已经成为一种有效的合成大环的方法.
2006年, 祝介平课题组[47]报道了利用Ugi/click串联反应合成14~16元大环的策略(Eq. 5).反应以正己醛、α-异腈-N-(丙炔)乙酰胺(18)和含叠氮的底物19为原料, 在甲苯中氯化铵的作用下80 ℃反应4 h得到Ugi产物.随后, 除去反应体系中的无机盐, 再在四氢呋喃中CuI和碱的作用下于室温发生环加成反应, 以45%的收率生成大环结构20.在这个简单的连续过程中, 同时形成了五个化学键, 并生成了一个大环和两个杂环, 且没有发现分子间的二聚产物.它的不足之处是需要化学计量的CuI和较低的反应浓度(c=0.001 mol/L).另外, 作者发现氯化铵的存在能使得Ugi反应的产率达到优秀. 2012年, Sureshbabu等[48]报道了利用连续的Ugi/click反应来合成三唑连接的环糖多肽类似物.
2015年, Kappe课题组[49]发展了基于连续流的通过Ugi反应/铜催化的click反应序列构建大环的策略(Scheme 8).与传统的方法不同的是, 溶于甲醇或乙腈的各个组分经各自的管路系统混合, 在一定温度下的铜圈中发生反应, 以良好的收率得到相应的产物.反应过程中的异腈是由伯吉斯试剂使相应的酰胺脱水现场生成.环加成中的叠氮化物也是通过溴化物前体和四丁基叠氮化胺的亲核取代得到.整个反应过程是连续的, 不需要任何中间体的分离, 从最初的原料到线性产物22或从线性产物到环多肽23都只需大约25 min.
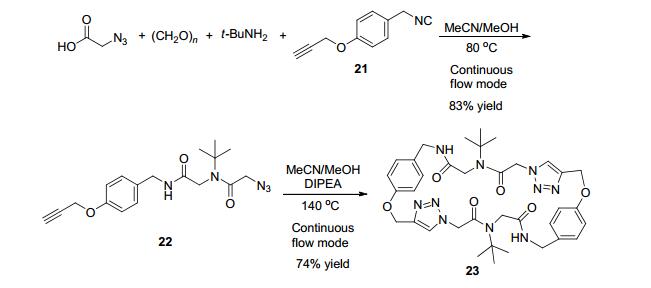 图式8
基于连续流手段合成环状多肽
Scheme8.
Synthesis of cyclic peptoids based on continuous flow
图式8
基于连续流手段合成环状多肽
Scheme8.
Synthesis of cyclic peptoids based on continuous flow
2 Ugi/Aldol反应
Aldol反应是在20世纪由Wurtz发现的, 它现在仍然是已知的通过简单原料构建碳碳键的最有效方法之一.
2012年, Hulme课题组[50]报道了连续的Ugi/aldol反应一锅法合成多取代杂环的方法.他们还用这样的方法合成了多取代杂双环化合物25.首先, 乙酰乙酸甲酯、α-羰基羧酸、胺和异腈反应生成Ugi产物, 然后在DMF中碱的作用下于180 ℃微波反应30 min发生环化反应, 以中等收率得到顺式并环的双环化合物(Scheme 9).
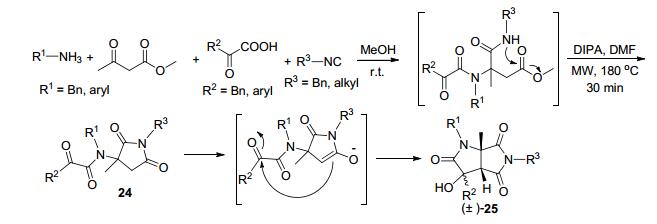 图式9
Ugi/aldol序列合成杂双环化合物
Scheme9.
Synthesis of bicyclic compounds through Ugi/aldol sequence
图式9
Ugi/aldol序列合成杂双环化合物
Scheme9.
Synthesis of bicyclic compounds through Ugi/aldol sequence
2013年, Hulme课题组[51]以邻氟苯胺为原料通过Ugi反应/分子内aldol/分子内亲核取代合成了三环化合物(Scheme 10).包括邻氟苯胺在内的起始原料在室温下搅拌过夜得Ugi产物26, 然后在干燥的DMF中以DIPEA为碱于120 ℃反应过夜, 即可构建三环骨架27.此方法的底物范围较宽, 三步的产率为中等, 适合于快速合成复杂的杂环分子.
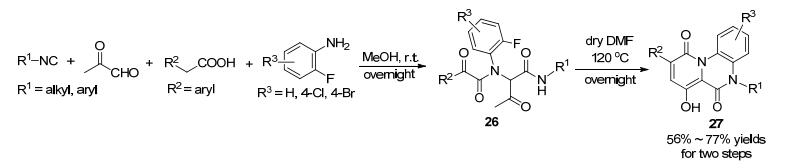 图式10
Ugi/aldol/SN2序列合成杂环
Scheme10.
Synthesis of heterocycles through Ugi/aldol/SN2 sequence
图式10
Ugi/aldol/SN2序列合成杂环
Scheme10.
Synthesis of heterocycles through Ugi/aldol/SN2 sequence
2015年丁明武课题组[52]以α, β-不饱和芳基羧Baylis-Hillman鏻盐28、α-羰基醛、胺和异腈为原料, 通过Ugi/aldol/hydrolysis反应序列, 实现了5, 6-二氢吡啶-2(1H)-酮化合物的一锅法合成(Scheme 11). Baylis-Hillman鏻盐可由α-甲基-β-芳基丙烯酸溴代后, 经三苯基膦处理得到.反应首先将各组分在甲醇中于室温反应24 h得到Ugi产物, 然后在固体碳酸钾的作用下于60 ℃经分子内aldol反应和水解反应以中等收率生成5, 6-二氢吡啶-2(1H)-酮化合物30.
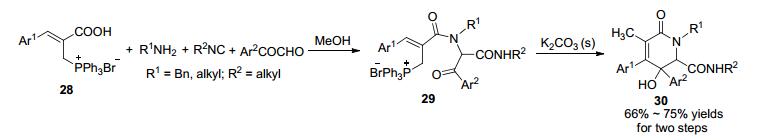 图式11
Ugi/aldol/hydrolysis序列合成5, 6-二氢吡啶-2(1H)-酮
Scheme11.
Synthesis of 5, 6-dihydropyridin-2(1H)-ones through Ugi/aldol/hydrolysis sequence
图式11
Ugi/aldol/hydrolysis序列合成5, 6-二氢吡啶-2(1H)-酮
Scheme11.
Synthesis of 5, 6-dihydropyridin-2(1H)-ones through Ugi/aldol/hydrolysis sequence
3 Ugi-Michael加成反应
Michael加成反应是有机合成领域一类非常重要的反应, 常用于C—C键和C—杂原子键的构建. 2010年, 杨震课题组[53]使用简单的原料, 通过连续的Ugi反应/分子内Michael加成反应一锅法非对映选择性地合成了基于香豆素的三环化合物(Eq. 6).香豆素-3-羧酸、吡啶醛、芳胺和异腈在甲醇中反应即可以以45%到82%的产率得到[3, 4-c]吡咯-3, 4-二酮31.在该反应中, 起始原料醛必须是邻位或对位的吡啶醛, 间位的吡啶醛不能发生反应.
Andrean等[54]利用微波促进的连续Ugi反应/Michael加成/氮杂-Michael加成反应合成了氮杂螺三环(Eq. 7).该反应溶剂是水, 不需添加剂.为了减少副反应和提高反应产率, 作者将反应分成了两个阶段:第一阶段, 原料在水中于70 ℃微波作用下反应1 h; 第二阶段, 温度升至190 ℃继续反应30 min, 最后以65%到90%的产率得到了氮杂螺三环化合物33.值得注意的是, 此种连续过程以良好至优秀的产率以及较好的非对映选择性产生了包括一个季碳中心在内的四个手性中心和六个连续的化学键.通过异腈的改变及后期的化学操作, 可以将这些化合物进一步多样化和功能化.
螺-2, 5-二酮哌嗪(spiro-2, 5-diketopiperazines, DKPs), 是一类具有生理活性的化合物.有报道称它们是特异性糖原磷酸化酶的抑制剂, 另外还和β-转角模拟物也有一定的相似性. Andrean等[55]报道了一种微波促进的连续Ugi反应/氮杂-Michael加成反应, 实现了螺-2, 5-二酮哌嗪(spiro-DKPs)的快速高效合成(Eq. 8).同样的, 该反应也是分为两个阶段进行的. 2012年Holzgrabe等[56]也利用连续Ugi反应/氮杂-Michael加成反应合成了此类化合物.
2016年, Srivastava等[57]利用连续的Ugi反应/环化/氮杂-Michael加成反应构建了氮杂螺环三环(Scheme 12). Ugi反应各原料组分在乙腈和水[V(CH3CN):V(H2O)=30:1]中于室温反应12 h, 接着与碘反应5~6 h生成去芳构化的产物37, 然后再在二氯乙烷中与过量浓硫酸于60 ℃反应2.5 h, 最后通过Suzuki偶联以65%的产率和99:1的dr值得到最终产物38.该方法以较高的区域选择性和非对映选择性生成了三个手性中心(包括一个季碳中心), 可用于合成多重官能团化的氮杂螺三环骨架结构.总的来说, 这种方法为多样化的生物小分子库的建立提供了有效途径, 还可以用于生物碱的全合成.
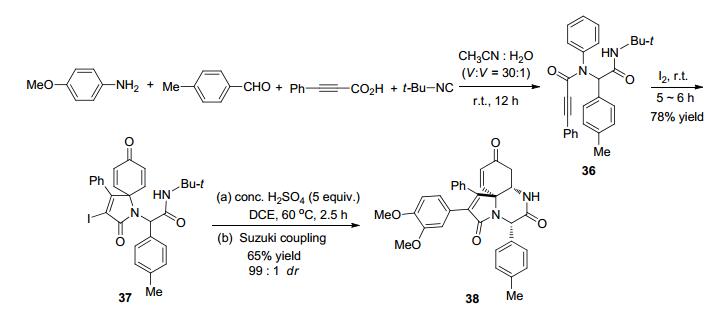 图式12
Ugi/Michael/交叉偶联序列合成氮杂螺三环
Scheme12.
Synthesis of azaspirofused tricyclic product through Ugi/Michael/cross-coupling sequence
图式12
Ugi/Michael/交叉偶联序列合成氮杂螺三环
Scheme12.
Synthesis of azaspirofused tricyclic product through Ugi/Michael/cross-coupling sequence
2016年, Van der Eycken课题组[58]报道了一种基于咪唑的快速、高效、绿色地构建杂三环的方法(Scheme 13).咪唑甲醛、炔胺、炔酸和异腈在甲醇中于室温反应24 h, 再升温至50 ℃反应24 h, 生成Michael加成产物40.然后在水中银的催化作用下120 ℃微波反应30 min, 以52%到93%的产率得到最终产物41.该方法可以快速地以简单易得的原料构建杂三环.另外, 该序列最后一步的反应介质是水, 符合绿色化学的发展理念.
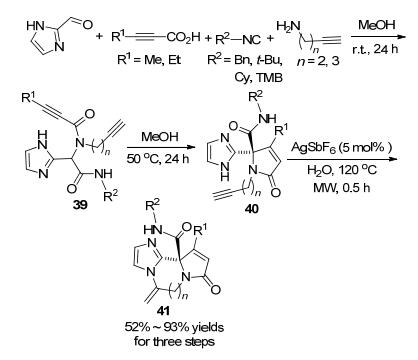 图式13
Ugi/Michael/银催化的环化序列
Scheme13.
Ugi/Michael/silver-catalyzed cyclization sequence
图式13
Ugi/Michael/银催化的环化序列
Scheme13.
Ugi/Michael/silver-catalyzed cyclization sequence
4 与Ugi反应串联的偶联反应
2006年, 祝介平课题组[59]首次利用串联的Ugi反应/Buchwald-Hartwig偶联来合成氧化吲哚(Scheme 14).起始原料先在甲醇中室温下反应, 然后再在甲苯和乙腈[V(甲苯):V(乙腈)=3:1]中催化量的钯络合物和碳酸钾的作用下于100 ℃微波反应1~2.5 h, 就可以以55%到99%的收率生成氧化吲哚43.其中, 反应第二步的(邻-碘苯基)-2-酰氨基乙酰胺42的分子内环化是在微波辅助下完成的.通过对比实验发现, 微波能显著加快反应速率和提高反应收率.这种将Ugi反应和Buchwald-Hartwig偶联结合的策略, 可以由简单的原料制备高度官能团化的氧化吲哚.
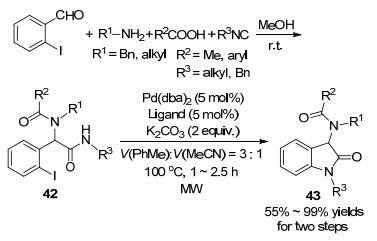 图式14
Ugi/Buchwald-Hartwig序列合成氧化吲哚
Scheme14.
Synthesis of oxindoles by Ugi/Buchwald-Hartwig sequence
图式14
Ugi/Buchwald-Hartwig序列合成氧化吲哚
Scheme14.
Synthesis of oxindoles by Ugi/Buchwald-Hartwig sequence
2014年, Van der Eycken课题组[60]报道了一种通过连续的Ugi反应/Pd(0) 催化的Buchwald-Hartwig偶联/ Michael加成反应来合成螺[二氢吲哚-3, 2'-吡咯]-2, 5'-二酮的策略(Scheme 15).起始原料先在甲醇中于室温反应得到Ugi产物, 再以Xantphos为配体、Pd(OAc)2为金属催化剂、碳酸铯为碱, 在甲苯中于120 ℃反应24 h可以以中等的产率得到最终产物44.碱、金属、配体对该反应的影响非常大, 稍微改变反应条件则反应的产率很低或不进行.炔酸可以被α, β-不饱和酸(例如肉桂酸或阿托酸)代替来制备多样化的螺环氧化吲哚.
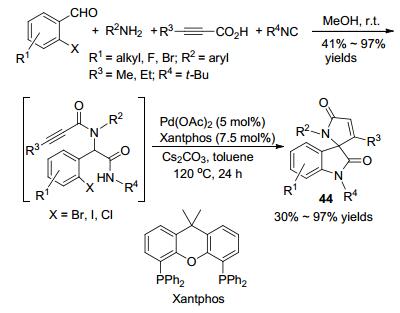 图式15
Ugi/Buchwald-Hartwig/Michael序列合成螺杂环
Scheme15.
Synthesis of spirocyclic heterocycles by Ugi/ Buchwald-Hartwig/Michael reaction
图式15
Ugi/Buchwald-Hartwig/Michael序列合成螺杂环
Scheme15.
Synthesis of spirocyclic heterocycles by Ugi/ Buchwald-Hartwig/Michael reaction
同年, Singh课题组[61]报道了类似的方法, 采用连续的Ugi/Heck/双键迁移反应一锅法地合成了1, 2-二氢苯并[b][1, 6]萘啶(Scheme 16). 3-氯喹啉-3-甲醛(45)、异腈、烯丙基胺和乙酸在甲醇中反应10~12 h生成Ugi产物46, 然后再以DMF为溶剂在催化量的醋酸钯和两倍量的醋酸钠的作用下, 在90 ℃下反应, 最后以良好的收率生成最终产物47.第二步的Heck反应是在DMF中进行的, 不需要外加配体.在其它溶剂(如DMSO、乙腈等)中或外加配体(如三苯基膦)的存在下反应的产率很低.
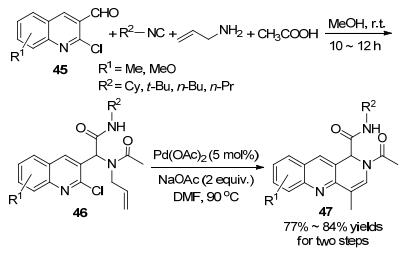 图式16
Ugi/Heck/双键迁移序列合成杂环
Scheme16.
Synthesis of heterocycles through Ugi/Heck/double bond migration sequence
图式16
Ugi/Heck/双键迁移序列合成杂环
Scheme16.
Synthesis of heterocycles through Ugi/Heck/double bond migration sequence
2015年, Van der Eycken课题组[62]报道了通过Ugi/还原Heck反应来合成苯并氮杂环化合物的策略.炔基羧酸、邻溴苯乙醛、异腈和胺在甲醇中反应通过Ugi反应生成炔丙酰胺(Scheme 17), 然后炔丙酰胺在微波的作用下反应生成3-苯并氮杂环化合物49.两步反应的产率均在中等或中等以上.作者还对该反应的底物适用性进行了考察, 邻卤代(Br、I)苯乙胺也可作为胺源参与本反应.
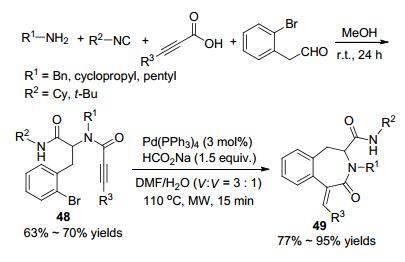 图式17
Ugi/还原Heck序列合成3-苯并氮杂化合物
Scheme17.
Synthesis of 3-benzazepine products through Ugi/reductive Heck sequence
图式17
Ugi/还原Heck序列合成3-苯并氮杂化合物
Scheme17.
Synthesis of 3-benzazepine products through Ugi/reductive Heck sequence
5 Ugi/Wittig反应
最近十几年, Ugi/Wittig反应序列被广泛应用于多取代杂环的合成[63~65]. 2004年, D mling等[66]首次利用Ugi/Honer-Wadsworth-Emmons连续反应以高效的汇聚式策略合成了多取代的吡咯烷酮和吡啶酮(Scheme 18).在甲醇中, 异腈、伯胺、芳基乙二醛和α-磷酸酯羧酸于室温下反应得Ugi产物50, 然后以THF为溶剂在LiCl和三乙胺的作用下发生Wittig反应, 最终以小于50%的总产率得到吡咯烷酮51.尽管这个反应的产率较低, 但其原料简单易得, 仍具有较大的潜在应用价值.
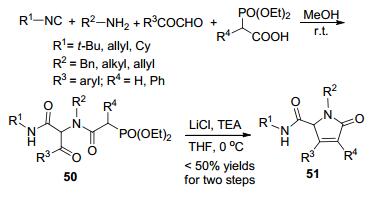 图式18
连续的Ugi/Wittig反应合成吡咯烷酮
Scheme18.
Synthesis of pyrrolidinones by tandem Ugi/Wittig reaction
图式18
连续的Ugi/Wittig反应合成吡咯烷酮
Scheme18.
Synthesis of pyrrolidinones by tandem Ugi/Wittig reaction
亚胺基膦烷最初是由Staudinger和Meyer制备, 是一类重要的氮杂-Wittig试剂, 可用于合成含氮杂环化合物. 2010年, Rezaei等[67]首次将(N-异氰亚氨基)三苯基膦应用于Ugi/氮杂-Wittig连续反应, 实现了一锅法构建1, 3, 4-噁二唑(Eq. 9).芳基醛、仲胺、芳基羧酸和(N-异氰亚氨基)三苯基膦(52)在二氯甲烷中于室温下反应2 h以93%的产率得到噁二唑53.该方法条件温和、产率较高, 可用于二取代的1, 3, 4-噁二唑的快速、高效合成.
2015年, 丁明武课题组[68]报道了以鏻盐为原料的Ugi/Wittig连续反应, 一锅法地合成了苯并氮杂环庚酮(Scheme 19).包括芳基羧酸鏻盐的起始原料在甲醇中于室温下反应, 然后再以三乙胺为碱在甲苯中回流, 最后生成了3H-2-苯并氧杂环庚烯-1-酮产物(55), 产率为52%至87%.邻甲基苯甲酸与N-溴代琥珀酰亚胺(NBS)溴化产生邻溴甲基苯甲酸, 然后再和三苯基膦反应可得鏻盐前体.鏻盐中间体在极性溶剂中碱的作用下容易分解, 故第二步反应是在甲苯中进行的.反应的底物适用范围比较宽并且原料简单易得, 这使得本策略可以用来制备多样化的苯并氮杂环.
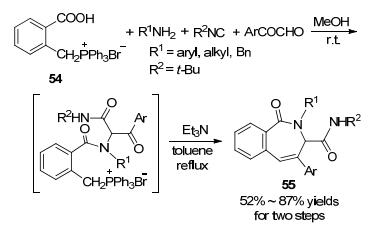 图式19
Ugi/Wittig序列合成苯并氮杂环庚酮
Scheme19.
Synthesis of benzoazepanones through Ugi/Wittig sequence
图式19
Ugi/Wittig序列合成苯并氮杂环庚酮
Scheme19.
Synthesis of benzoazepanones through Ugi/Wittig sequence
同年, Welsch等[69]报道了通过Ugi/Staudinger/Wittig反应序列来合成取代的咪唑啉(Scheme 20)的策略.叠氮基乙胺、羧酸、苄基异腈和醛在甲醇中于室温下反应以64%~90%的产率生成了Ugi产物, 然后其在干燥的甲苯中三苯基膦的作用下发生Staudinger反应, 最后在150 ℃下进行微波反应以72%~99%的产率得到取代的咪唑啉57.该反应也可以以2-氨基乙-1-醇为原料, 并在Ugi反应之后通过取代发应引入叠氮基.若用溴乙胺, 则第一步的Ugi反应不能进行.
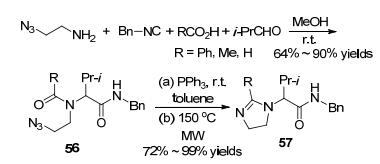 图式20
Ugi/Staudinger/Wittig序列合成咪唑啉
Scheme20.
Synthesis of imidazolines by Ugi/Staudinger/Wittig sequence
图式20
Ugi/Staudinger/Wittig序列合成咪唑啉
Scheme20.
Synthesis of imidazolines by Ugi/Staudinger/Wittig sequence
2016年, 丁明武课题组[70]采用类似的策略合成了多取代苯并咪唑(Scheme 21).他们直接以2-叠氮基苯胺为原料在室温下反应24~48 h, 以中等产率得到了Ugi产物58, 然后在二氯甲烷中还原剂三苯基膦的作用下生成亚氨基磷烷中间体, 最后通过氮杂-Wittig反应以67%~92%的产率生成多取代的苯并咪唑59.反应的底物适用性较广.邻位叠氮化的芳胺, 经过该反应序列即可高效的合成多取代苯并咪唑.
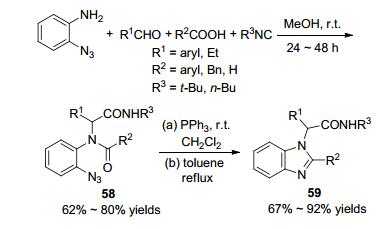 图式21
Ugi/Staudinger/Wittig序列合成多取代苯并咪唑
Scheme21.
Synthesis of multisubstituted benzimidazoles by Ugi/Staudinger/Wittig sequence
图式21
Ugi/Staudinger/Wittig序列合成多取代苯并咪唑
Scheme21.
Synthesis of multisubstituted benzimidazoles by Ugi/Staudinger/Wittig sequence
6 其它post-Ugi反应
金作为软的Lewis酸可以和炔、烯烃及丙二烯形成金络合物, 使其被活化而可以被亲核试剂进攻.均相金催化的碳环化和杂环化已经成为极其活跃的研究领域.金催化的炔和芳环或芳杂环的分子内环化反应, 为构建具有生物活性的化合物提供了新的合成途径[71~73]. 2013年, Van der Eycken课题组[74]报道了在Ugi反应基础上的Au(Ⅰ)催化的环化反应(Scheme 22).咪唑甲醛60、炔酸、异腈和胺在甲醇中于室温反应生成Ugi产物61, 然后以氯仿为溶剂在Au(PPh3)Cl或AgBF4的作用下发生环化反应, 以75%至98%的产率生成咪唑并[1, 4]二氮杂-7-酮62.炔基上大位阻的基团降低了环化反应的速率, 升高温度和增加催化剂用量才能使反应顺利进行.虽然AgBF4也可以催化该反应, 但是反应时间较长, 需要24~30 h, 而Au(PPh3)Cl催化的反应仅需4 h.咪唑并二氮杂酮常用作构成药物分子的骨架.它是已商品化的抗肿瘤药喷司他丁(pentostanin)、抗病毒药助间型霉素(coformycin)和抗焦虑药溴它西尼(bretazenil)的重要合成中间体.
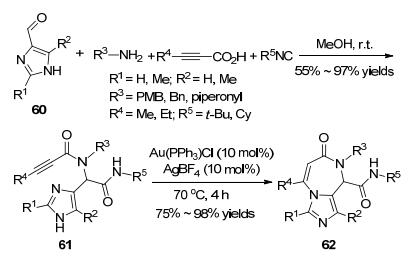 图式22
Ugi/金催化的环化序列
Scheme22.
Ugi/Gold-catalyzed cyclization sequence
图式22
Ugi/金催化的环化序列
Scheme22.
Ugi/Gold-catalyzed cyclization sequence
Bischler-Napieralski反应是指β-芳基乙基酰胺在氯代试剂的作用下发生分子内亲电取代, 环合生成二氢异喹啉类化合物的反应.该反应是Bischler和Napieral-ski[75]于1893年在研究生物碱时发现的. Bischler-Napieralski反应可用于异喹啉类药物及其它杂环的合成. 2012年, Ho课题组[76]在合成用于治疗精神分裂症的二氢咪唑异喹啉衍生物时, 采用了Ugi/Bischler-Na-pieralski串联反应. 2014年, Vece课题组[77]利用Ugi/Bischler-Napieralski连续过程合成了氮杂环(Scheme 23).起始原料醛、胺、羧酸和3-(2-异氰基乙基)-吲哚(63)在甲醇中于室温下反应得到Ugi产物64, 然后于甲苯中在POCl3的作用下继续反应, 最后以70%到94%的产率生成含咪唑鎓盐的杂环化合物65.当R2为氢原子时, 得到的是咪唑产物而不是其鎓盐.
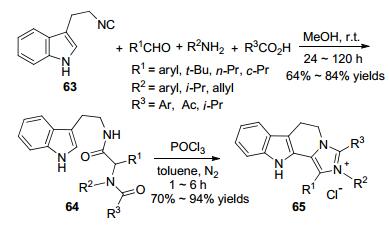 图式23
Ugi/Bischler-Napieralski环化序列
Scheme23.
Ugi/Bischler-Napieralski cyclization sequence
图式23
Ugi/Bischler-Napieralski环化序列
Scheme23.
Ugi/Bischler-Napieralski cyclization sequence
Kazmaier等[78]通过立体选择性的Ugi/RCM串联反应来构建环多肽(Scheme 24).异氰基乙酸烯丙酯、(S)-1-(3-甲氧基苯基)乙-1-胺、(S)-氨基酸和醛在三氟乙醇中反应得到Ugi产物67, 然后在第一代Grubbs催化剂的作用下以37%~51%的产率关环得到16元大环多肽68.第一步Ugi反应的非对映选择性对温度有很大的依赖性, 在-30 ℃时, dr值为9:5;而在室温或0 ℃时, dr值则很低.此外, 反应的产率受溶剂的影响较大, 只有在三氟乙醇作溶剂时产率才可以达到良好.
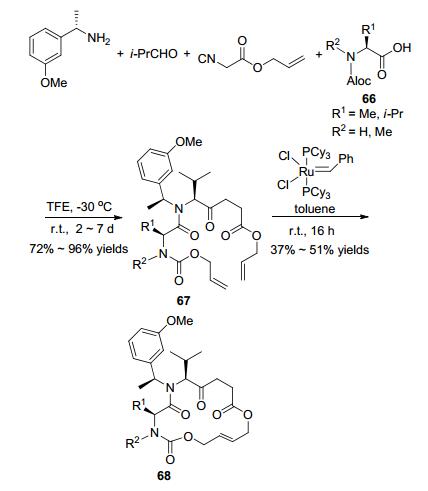 图式24
Ugi/RCM序列构建大环
Scheme24.
Ugi/RCM sequence to construct macrocycles
图式24
Ugi/RCM序列构建大环
Scheme24.
Ugi/RCM sequence to construct macrocycles
2012年, Banfi课题组[79]通过Mitsunobu反应将Ugi酰胺进一步环化, 合成了苯并噁嗪酮(Scheme 25).邻羟基苯胺、α羟基羧酸、醛和异腈在异丙醇中反应可以以57%~81%的产率生成Ugi酰胺69, 然后在二氯甲烷中三苯基膦和DEAD的作用下发生环化生成苯并噁嗪酮70, 产率为80%~95%.在第一步中, 脂肪醛参与的Ugi反应产率低于芳香醛.为了抑制传统溶剂甲醇与乙醇酸的酯化反应, 他们选用了异丙醇作溶剂.作者还发现反应以分步进行或“一锅法”进行都是以相同的产率生成苯并噁嗪酮.
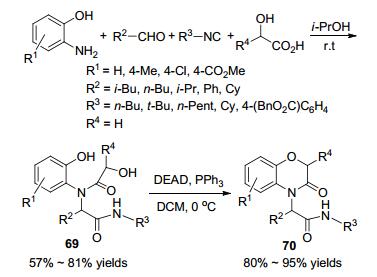 图式25
Ugi/Mitsunobu序列合成苯并噁嗪酮
Scheme25.
Synthesis of benzoxazinones by Ugi/Mitsunobu sequence
图式25
Ugi/Mitsunobu序列合成苯并噁嗪酮
Scheme25.
Synthesis of benzoxazinones by Ugi/Mitsunobu sequence
6 总结与展望
Ugi反应具有操作简便、原子经济性好、收率高等特点. Post-Ugi反应在Ugi反应原有的基础上拓宽了其应用, 可广泛应用于结构多样的分子特别是含氮杂环化合物的合成.除了文中叙述的反应以外, 还有Ugi/ Ullmann反应[80]、Ugi/Diels-Alder反应[81, 82]、Ugi/Pictet-Spengler反应[83]等也有报道.随着人们对于杂环化合物的日益关注和组合化学的不断发展, post-Ugi反应将被人们进一步研究和发掘, 进而探索一些新颖、高效的杂环合成方法.
-
-
[1]
Tzitzikas, T. Z.; Chandgude, A. L.; Dömling, A. Chem. Rec. 2015, 15, 981. doi: 10.1002/tcr.v15.5
-
[2]
Ruijter, E.; Scheffelaar, R.; Orru, R. V. A. Angew. Chem., Int. Ed. 2011, 50, 6234. doi: 10.1002/anie.201006515
-
[3]
Zhu, J. P.; Wang, Q.; Wang, M. X. Multicomponent Reaction in Organic Synthesis, Wiley-VCH, Weinheim, 2015.
-
[4]
Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083. doi: 10.1021/cr100233r
-
[5]
Benjamin, H. R.; Zaretsky, S.; Rai, V.; Yudin, K. A. Chem. Rev. 2014, 114, 8323. doi: 10.1021/cr400615v
-
[6]
Tang, Z.; Liu, Z.; An, Y.; Jiang, R.; Zhang, X.; Li, C.; Jia. X. J.Org. Chem. 2016, 81, 9158. doi: 10.1021/acs.joc.6b01711
-
[7]
Ugi, I.; Meyr, R.; Fetzer, U. Angew. Chem. 1959, 71, 386.
-
[8]
Váradi, A.; Palmer, T. C.; Dardashti, R. N.; Majumdar, S. Molecules 2016, 21, 19.
-
[9]
胡汉宁, 黎安玲, 张瀚匀, 石德清, 有机化学, 2015, 35, 2162. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345200.shtmlHu, H.; Li, A.; Zhang, H.; Shi, D. Chin. J. Org. Chem. 2015, 35, 2162(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345200.shtml
-
[10]
詹益周, 王宝雷, 张丽媛, 张燕, 张晓, 李正名, 宋海斌, 化学学报, 2015, 73, 1173. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344768.shtmlZhan, Y.; Wang, B.; Zhang, L.; Zhang, Y.; Zhang, X.; Li, Z.; Song, H. Acta Chim. Sinica 2015, 73, 1173(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344768.shtml
-
[11]
肖立伟, 彭晓霞, 周秋香, 寇伟, 时亚茹, 有机化学, 2015, 35, 1204. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344851.shtmlXiao, L.; Peng, X.; Zhou, Q.; Kou, W.; Shi, Y. Chin. J. Org. Chem. 2015, 35, 1204(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344851.shtml
-
[12]
Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168. doi: 10.1002/(ISSN)1521-3773
-
[13]
Sadjadi, S.; Heravi, M. M.; Nazarib, N. RSC Adv. 2016, 6, 53203. doi: 10.1039/C6RA02143C
-
[14]
Haji, M. Beilstein J. Org. Chem. 2016, 12, 1269. doi: 10.3762/bjoc.12.121
-
[15]
王也铭, 刘兆洪, 唐春霖, 毕锡和, 分子科学学报, 2016, 32, 1. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345257.shtmlWang, Y.; Liu, Z.; Tang, C.; Bi, X. J. Mol. Sci. 2016, 32, 1(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345257.shtml
-
[16]
张钊瑞, 郑晓霖, 郭长彬, 有机化学, 2016, 36, 1241. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345415.shtmlZhang, Z.; Zheng, X.; Guo, C. Chin. J. Org. Chem. 2016, 36, 1241(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345415.shtml
-
[17]
Ugi, I. Angew. Chem. 1961, 74, 9.
-
[18]
Nixey, T.; Kelly, M.; Hulme, C. Tetrahedron Lett. 2000, 41, 8729. doi: 10.1016/S0040-4039(00)01563-X
-
[19]
Nixey, T.; Kelly, M.; Semin, D.; Hulme, C. Tetrahedron Lett. 2002, 43, 3681. doi: 10.1016/S0040-4039(02)00636-6
-
[20]
Nayak, M.; Batra, S. Tetrahedron Lett. 2010, 51, 510. doi: 10.1016/j.tetlet.2009.11.051
-
[21]
Nixey, T.; Kelly, M.; Hulme, C. Tetrahedron Lett. 2000, 41, 8729. doi: 10.1016/S0040-4039(00)01563-X
-
[22]
Kalinski, C.; Umkehrer, M.; Gonnard, S.; Jager, N.; Ross, G.; Hillerb, W. Tetrahedron Lett. 2006, 47, 2041. doi: 10.1016/j.tetlet.2006.01.027
-
[23]
Yerande, S. G.; Newase, K. M.; Singh, B.; Boltjes, A.; Dömling, A. Tetrahedron Lett. 2014, 55, 3263. doi: 10.1016/j.tetlet.2014.04.040
-
[24]
Borisov, R. S.; Polyakov, A. I.; Medvedeva, L. A.; Khrustalev, V. N.; Guranova, N. I.; Voskressensky, L. G. Org. Lett. 2010, 12, 3894. doi: 10.1021/ol101590w
-
[25]
Shmatova, O. I.; Nenajdenko, V. G. Eur. J. Org. Chem. 2013, 6397.
-
[26]
Shmatova, O. I.; Nenajdenko, V. G. J. Org. Chem. 2013, 78, 9214. doi: 10.1021/jo401428q
-
[27]
Mikaimi, K.; Lautens, M. New Frontiers in Asymmetric Catalysis, Wiley, New York, 2007.
-
[28]
Ugi, I.; Rosendahl, F. K. Justus Liebigs Ann. Chem. 1963, 666, 65. doi: 10.1002/(ISSN)1099-0690
-
[29]
Shevchenko, N. E.; Nenajdenko, V. G.; Röschenthaler, G.-V. J. Fluorine Chem. 2008, 129, 390. doi: 10.1016/j.jfluchem.2008.01.013
-
[30]
Nenajdenko, V. G.; Zakurdaev, E. P.; Prusov, E. V.; Balenkova, E. S. Tetrahedron 2004, 60, 11719. doi: 10.1016/j.tet.2004.10.006
-
[31]
Kroon, E.; Kurpiewska, K.; Kalinowska-Tłusćik, J.; Dömling, A. Org. Lett. 2016, 18, 4762. doi: 10.1021/acs.orglett.6b01826
-
[32]
Nutt, R. F.; Joullié, M. M. J. Am. Chem. Soc. 1982, 104, 5852. doi: 10.1021/ja00385a080
-
[33]
Flanagan, D. M.; Joullié, M. M. Synth. Commun. 1989, 19, 1. doi: 10.1080/00397918908050946
-
[34]
Katsuyama, A.; Matsuda, A.; Ichikawa, S. Org. Lett. 2016, 18, 2552 doi: 10.1021/acs.orglett.6b00827
-
[35]
Banfi, L.; Basso, A.; Guanti, G.; Riva, R. Tetrahedron Lett. 2004, 45, 6637. doi: 10.1016/j.tetlet.2004.07.015
-
[36]
Hua, D. H.; Miao, S. W.; Bharathi, S. N.; Katsuhira, T.; Bravo, A. A. J. Org. Chem. 1990, 55, 3682. doi: 10.1021/jo00298a062
-
[37]
Golubev, P.; Bakulina, O.; Darin, D.; Krasavin, M. Eur. J. Org. Chem. 2016, 3969.
-
[38]
Morana, F.; Basso, A.; Bella, M.; Riva, R.; Banfi, L. Adv. Synth. Catal. 2012, 354, 2199. doi: 10.1002/adsc.v354.11/12
-
[39]
Chapman, T. M.; Davies, I. G.; Gu, B.; Block, T. M.; Scopes, D. I. C.; Hay, P. A.; Courtney, S. M.; McNeill, L. A.; Schofield, C. J.; Davis, B. G. J. Am. Chem. Soc. 2005, 127, 506. doi: 10.1021/ja043924l
-
[40]
Znabet, A.; Ruijter, E.; de Kanter, F. J. J.; Kohler, V.; Helliwell, M.; Turner, N. J.; Orru, R. V. A. Angew. Chem., Int. Ed. 2010, 49, 5289. doi: 10.1002/anie.v49:31
-
[41]
刘学超, 博士论文, 南京大学, 南京, 2011.Li, X.-C. Ph.D. Dissertation, Nanjing University, Nanjing, 2011(in Chinese).
-
[42]
Nenajdenko, V. G.; Gulevich, A. V.; Sokolova, N. V.; Mironov, A. V.; Balenkova. E. S. Eur. J. Org. Chem. 2010, 1445.
-
[43]
Vorobyeva, D. V.; Sokolova, N. V.; Nenajdenko, V. G., Peregudov, A. S.; Osipov, S. N. Tetrahedron 2012, 68, 872. doi: 10.1016/j.tet.2011.11.037
-
[44]
Pramitha, P.; Bahulayan, D. Bioorg. Med. Chem. Lett. 2012, 22, 2598. doi: 10.1016/j.bmcl.2012.01.111
-
[45]
Niu, T. F.; Gu, L.; Yi, W. B.; Cai, C. ACS Comb. Sci. 2012, 14, 309. doi: 10.1021/co3000117
-
[46]
Shin, S. B. Y.; Yoo, B.; Todaro, L. J.; Kirshenbaum, K. J. Am. Chem. Soc. 2007, 129, 3218. doi: 10.1021/ja066960o
-
[47]
Pirali, T.; Tron, G. C.; Zhu, J. Org. Lett. 2006, 8, 4145. doi: 10.1021/ol061782p
-
[48]
Samarasimhareddy, M.; Hemantha, H. P.; Sureshbabu, V. V. Tetrahedron Lett. 2012, 53, 3104. doi: 10.1016/j.tetlet.2012.04.034
-
[49]
Salvador, C. E. M.; Pieber, B.; Neu, P. M.; Torvisco, A.; Andrade, C. K. Z.; Kappe, C. O. J. Org. Chem. 2015, 80, 4590. doi: 10.1021/acs.joc.5b00445
-
[50]
Xu, Z.; Moliner, F. D.; Cappelli, A. P.; Hulme, C. Angew. Chem., Int. Ed. 2012, 51, 8037. doi: 10.1002/anie.v51.32
-
[51]
Xu, Z.; Moliner, F. D.; Cappelli, A. P.; Hulme, C. Org. Lett. 2013, 15, 2738. doi: 10.1021/ol401068u
-
[52]
Zeng, X.; Wang, H.; Ding, M. Org. Lett. 2015, 17, 2234. doi: 10.1021/acs.orglett.5b00849
-
[53]
Che, C.; Li, S.; Jiang, X.; Quan, J.; Lin, S.; Yang, Z. Org. Lett. 2010, 20, 4682.
-
[54]
Santra, S.; Andreana, P. R. Angew. Chem., Int. Ed. 2011, 50, 9418. doi: 10.1002/anie.v50.40
-
[55]
Santra, S.; Andreana, P. R. J. Org. Chem. 2011, 76, 2261. doi: 10.1021/jo102305q
-
[56]
Hartung, A.; Seufert, F.; Berges, C.; Gessner. V. H.; Holzgrabe, U. Molecules 2012, 17, 14685. doi: 10.3390/molecules171214685
-
[57]
Yugandhar, D.; Kuriakose, S.; Nanubolu, J. B.; Srivastava, A. K. Org. Lett. 2016, 18, 1040. doi: 10.1021/acs.orglett.6b00164
-
[58]
Li, Z.; Zhao, Y.; Tian, G.; Yi, H.; Song, G.; Meerveltc, L. V.; Eycken, E. V. V. RSC Adv. 2016, 6, 103601. doi: 10.1039/C6RA23180B
-
[59]
Bonnaterre, F.; Bois-Choussy, M.; Zhu, J. Org. Lett. 2006, 19, 4351.
-
[60]
Sharma, N.; Li, Z.; Sharma, U. K.; Van der Eycken, E. V. Org. Lett. 2014, 16, 3884. doi: 10.1021/ol5019079
-
[61]
Asthana, M.; Sharma, N.; Singh, M. R. Tetrahedron 2014, 70, 7996. doi: 10.1016/j.tet.2014.08.046
-
[62]
Peshkov, A. A.; Peshkov, V. A.; Pereshivko, O. P.; Van der Eycken, E. V. Tetrahedron 2015, 71, 3863. doi: 10.1016/j.tet.2015.04.022
-
[63]
He, P.; Nie, Y. B.; Wu, J.; Ding, M. W. Org. Biomol. Chem. 2011, 9, 1429. doi: 10.1039/c0ob00855a
-
[64]
Zhong, Y.; Wang, L.; Ding, M.-W. Tetrahedron 2011, 67, 3714. doi: 10.1016/j.tet.2011.03.056
-
[65]
Duan, Z.; Gao, Y.; Yuan, D.; Ding, M. W. Synlett 2015, 26, 2598. doi: 10.1055/s-00000083
-
[66]
Beck, B.; Picard, A.; Herdtweck, E.; Dömling, A. Org. Lett. 2004, 1, 39.
-
[67]
Ramazani, A.; Rezaei, A. Org. Lett. 2010, 12, 2852. doi: 10.1021/ol100931q
-
[68]
Wang, L.; Ren, Z.; Ding, M. J. Org. Chem. 2015, 80, 641. doi: 10.1021/jo502275f
-
[69]
Welsch, S. J.; Umkehrer, M.; Kalinski, C.; Ross, G.; Burdack, C.; Kolb, J.; Wild, M.; Ehrlich, A.; Wessjohann, L. A. Tetrahedron Lett. 2015, 56, 1025. doi: 10.1016/j.tetlet.2015.01.043
-
[70]
Yan, Y.-M.; Gao, Y.; Ding, M. W. Tetrahedron 2016, 72, 5548. doi: 10.1016/j.tet.2016.07.048
-
[71]
Furstner, A. Chem. Soc. Rev. 2009, 38, 3208. doi: 10.1039/b816696j
-
[72]
Bandini, M.; Bottoni, A.; Chiarucci, M.; Cera, G.; Miscione, G. P. J. Am. Chem. Soc. 2012, 134, 20690. doi: 10.1021/ja3086774
-
[73]
Vachhani, D. D.; Mehta, V. P.; Modha, S. G.; Van Hecke, K.; Van Meervelt, L.; Van der Eycken, E. V. Adv. Synth. Catal. 2012, 354, 1593. doi: 10.1002/adsc.201100881
-
[74]
Kumar, A.; L. Z.; Sharma, S. K.; Parmar, V. S.; Van der Eycken, E. V. Org. Lett. 2013, 8, 1874.
-
[75]
Bischler, A.; Napieralski, B. Chem. Ber. 1893, 26, 1903. doi: 10.1002/(ISSN)1099-0682
-
[76]
Ho, G. D.; Seganish, W. M.; Bercovici, A.; Tulshian, D.; Greenlee, W. J.; Van Rijn, R.; Hruza, A.; Xiao, L.; Rindgen, D.; Mullins, D.; Guzzi, M.; Zhang, X.; Bleickardt, C.; Hodgson, R. Bioorg. Med. Chem. Lett. 2012, 22, 2585. doi: 10.1016/j.bmcl.2012.01.113
-
[77]
Silvani, A.; Lesma, G.; Crippa, S.; Vece, V. Tetrahedron 2014, 70, 3994. doi: 10.1016/j.tet.2014.04.081
-
[78]
Hebach, C.; Kazmaier, U. Chem. Commun. 2003, 596.
-
[79]
Banfi, L.; Basso, A.; Giardini, L.; Riva, R.; Rocca, V.; Guanti, G. Eur. J. Org. Chem. 2011, 100.
-
[80]
Shi, J.; Wu, J.; Cui, C.; Dai, W.-M. J.Org. Chem. 2016, 81, 10392. doi: 10.1021/acs.joc.6b01398
-
[81]
Banfi, L.; Basso, A.; Guanti, G.; Lecinska, P.; Riva, R. Org. Biomol. Chem. 2006, 4, 4236. doi: 10.1039/b613056a
-
[82]
Cheng, G.; He, X.; Tian, L.; Chen, J.; Li, C.; Jia, X.; Li, J. J. Org. Chem. 2015, 80, 11100. doi: 10.1021/acs.joc.5b01724
-
[83]
Liu, H.; Domling, A. J. Org. Chem. 2009, 74, 6895. doi: 10.1021/jo900986z
-
[1]
-

图式2 Azido-Ugi反应合成四氮唑及其作为有机小分子催化剂的应用
Scheme 2 Synthesis of tetrazole-derived heterocycles via azido-Ugi reaction and its application as organocatalyst

图式3 可切断的β-腈基异腈应用于Ugi反应
Scheme 3 Application of cleavable β-cyanoethyl isocyanide in Ugi reaction

图式7 一锅法合成三唑修饰的肽类似物
Scheme 7 Synthesis of triazole-modified peptidomimetics through one-pot strategy

图式9 Ugi/aldol序列合成杂双环化合物
Scheme 9 Synthesis of bicyclic compounds through Ugi/aldol sequence

图式10 Ugi/aldol/SN2序列合成杂环
Scheme 10 Synthesis of heterocycles through Ugi/aldol/SN2 sequence

图式11 Ugi/aldol/hydrolysis序列合成5, 6-二氢吡啶-2(1H)-酮
Scheme 11 Synthesis of 5, 6-dihydropyridin-2(1H)-ones through Ugi/aldol/hydrolysis sequence

图式12 Ugi/Michael/交叉偶联序列合成氮杂螺三环
Scheme 12 Synthesis of azaspirofused tricyclic product through Ugi/Michael/cross-coupling sequence

图式14 Ugi/Buchwald-Hartwig序列合成氧化吲哚
Scheme 14 Synthesis of oxindoles by Ugi/Buchwald-Hartwig sequence

图式15 Ugi/Buchwald-Hartwig/Michael序列合成螺杂环
Scheme 15 Synthesis of spirocyclic heterocycles by Ugi/ Buchwald-Hartwig/Michael reaction

图式16 Ugi/Heck/双键迁移序列合成杂环
Scheme 16 Synthesis of heterocycles through Ugi/Heck/double bond migration sequence

图式17 Ugi/还原Heck序列合成3-苯并氮杂化合物
Scheme 17 Synthesis of 3-benzazepine products through Ugi/reductive Heck sequence

图式18 连续的Ugi/Wittig反应合成吡咯烷酮
Scheme 18 Synthesis of pyrrolidinones by tandem Ugi/Wittig reaction

图式19 Ugi/Wittig序列合成苯并氮杂环庚酮
Scheme 19 Synthesis of benzoazepanones through Ugi/Wittig sequence

图式20 Ugi/Staudinger/Wittig序列合成咪唑啉
Scheme 20 Synthesis of imidazolines by Ugi/Staudinger/Wittig sequence

图式21 Ugi/Staudinger/Wittig序列合成多取代苯并咪唑
Scheme 21 Synthesis of multisubstituted benzimidazoles by Ugi/Staudinger/Wittig sequence

图式23 Ugi/Bischler-Napieralski环化序列
Scheme 23 Ugi/Bischler-Napieralski cyclization sequence
-


 下载:
下载:

























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 69
- 文章访问数: 7804
- HTML全文浏览量: 2727
 下载:
下载:
