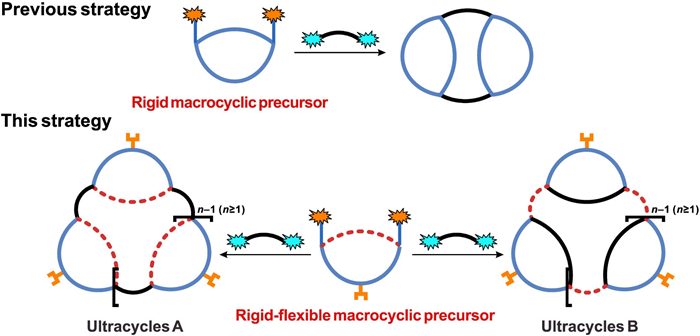
Scheme 1.
Construction of ultracycles using macrocyclic precursor.
Ultracycles consisting of macrocycles
Wen-Hui Mi , Teng-Yu Huang , Yu-Fei Ao , Xu-Dong Wang , Qi-Qiang Wang , De-Xian Wang

Macrocycles are common in nature and contribute one of the major driving forces to facilitate rapid progress of supramolecular chemistry and beyond [1-4]. In literature there are a few very large macrocycles that usually consist of > 50 ring atoms and show size of nanoscale, which were termed as ultracycles [5]. Growing interests have been paid to ultracycles as they represent the border area between molecular and nanoscale dimensions, and play important roles in natural process (such as macrolides or cyclic lipids), host‒guest chemistry and molecular machines (such as crown ethers, cyclophanes and calixarenes) [5, 6]. While most of the synthesized ultracycles were constructed using simple repeating units [7-15], very large macrocycles consisting of smaller macrocycles as the building units are largely underexplored [16-25]. Such peculiar macrocycle-containing ultracycles are expected to facilitate high association efficiency and selectivity towards guests arising from the cooperativity of the convergent macrocyclic elements [6, 26-28].
Despite the structural sophistication and potential function, the macrocycle-containing ultracycles are scarcely accessible in isolated form [5, 29]. The most successful strategy is template-directed synthesis of porphyrin-containing ultracycles which afforded very large macrocycles such as a 50-porphyrin ring with diameter of 21 nm [30]. In many cases the synthetic method relied on connection of macrocyclic elements (or macrocyclic precursor) with reactive linkages, which usually generated a mixture of ultracyclic products with different sizes, and hence lower chemical yields for individual ultracycles [17]. The shape persistency of the macrocyclic precursor would affect the size distribution of the products. For example, reaction of a shape-persistent of dichloro-oxacalix[2]arene[2]triazine with resorcinol or diamine linkers mainly produced bis-macrocycles (2 + 2) (Scheme 1) [31], while release of the conformation constrain engendered the formation of larger macrocycles (3 + 3 or 4 + 4) [28]. We envisioned the diversity of the ultracycles could be enriched through reversible cleavage and formation of aryl C‒O bonds: the direct assembly of macrocyclic precursor and linker would result in macrocycle A whereas aryl C‒O bond cleavage followed by intra- or inter-molecular C‒O bond formation afforded the rearranged macrocycle B (Scheme 1). Herein macrocyclic precursor bearing functional groups were designed combining the shape-persistent aromatic trimeric moiety to provide convergent reaction sites and glycol linkers to introduce local flexibility. Such rigid‒flexible macrocyclic precursor enables the diversity-oriented synthesis of functional ultracycles. Anion recognition properties of the isolated ultracycles are also documented.
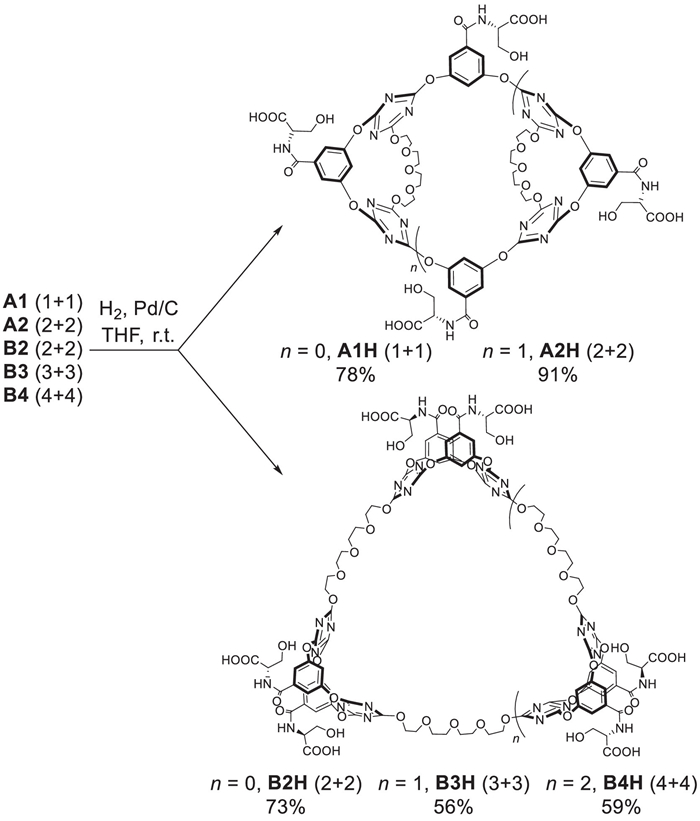
For synthesis of the ultracycles, we initially prepared the macrocyclic precursor 2 starting from cyanuric chloride, 3,6,9-trioxaundecane-1,11-diol and serine derivative substituted diphenol compound 1 in two steps (Schemes S1 and S2 in Supporting information). One-pot macrocyclization reaction between macrocycle 2 and monomer 1 was performed in presence of bases (Table 1). The type and amount of the bases were found to significantly influence the macrocyclization outcome. For example, the use of K2CO3 and Cs2CO3 produced macrocycles only in very low yields (entries 1 and 2). No isolated macrocycles could be obtained when 2 equiv. CsF was applied (entry 3). Pleasantly, increasing the amount of CsF to 4 equiv. afforded 1 + 1 (A1) and 2 + 2 (A2) macrocycle A, and 2 + 2 (B2) macrocycle B in total yield of 19.2% (entry 4). When the amount of CsF was increased to 6 equiv. only macrocycles B were produced, and in addition to the main product of 2 + 2 (B2), larger 3 + 3 (B3) and 4 + 4 (B4) ultracycles were also isolated in 4.0% and 1.1% yields, respectively (entry 5). The use of 8 equiv. CsF could accelerate the reaction and give products of macrocycle B with increased total chemical yield (entry 6). Further increase of CsF to 12 equiv. was detrimental to the total chemical yields of macrocycles B, probably due to the increase of byproducts in presence of excessive base (entry 7). In comparison with the non-substituted macrocyclic precursor which facilitated equal chances for dynamic and thermodynamic products when 4 and 8 equiv. CsF were applied respectively [31], the existence of serine derivative substituents on the precursor and monomer seems to favor the thermodynamic products (macrocycle B). As macrocycle A is the "normal" product from starting reagents, the "abnormal" product macrocycles B are possibly formed through a rearrangement pathway from macrocycle A. Such assumption could be confirmed by 1H NMR monitoring of A2 in presence of 10 equiv. CsF, from which formation of "abnormal" product B2, B3, B4 were clearly observed with 47% yields after 1 h (Fig. S6 in Supporting information). The proposed rearrangement mechanism could involve the cleavage of the dynamic covalent triazine‒O‒aryl bond in presence of nucleophile and the sequential formation of new triazine‒O‒Ar bond to furnish the thermodynamic macrocycles (Schemes S7 and S8 in Supporting information) [28].
As the resulting macrocycles contain anion binding sites (hydrogen bond donors and electron-deficient aromatics) and cation binding sites (glycol chain), they might show ability to bind large-size anions or alkali cations. Hence, we chose several organic anion salts to investigate the template effect on macrocyclization reactions (entries 8‒14). In presence of 4.0 equiv. CsF, the addition of mesylate, disulfonates, and trisulfonate as sodium salts could slightly increase the total yields of macrocycles. Different cations such as sodium or potassium hardly influence the reactions (entries 8 and 9). In the case of benzene-1,3,5-trimethanesulfonate, the use of ammonium was detrimental for macrocycle A, but favored the formation of macrocycles B (entry 14). These results suggested that the template effects mainly come from anions. The macrocyclic compounds were deprotected using Pd/C and H2 as reduction reagents to afford serine-substituted macrocycles A1H‒A2H and B2H‒B4H (Scheme 2). The synthesized macrocycles were fully characterized by spectrometric and elemental analysis (Supporting information).
We took B2H which contains two macrocyclic units as a representative example to investigate the anion recognition towards a series of dicarboxylates including oxalate (C22−), malonate (C32−), succinate (C42−), glutarate (C52−), adipate (C62−), heptanedioate (C72−) and octanedioate (C82−) (as tetrabutylammonium salts) by means of 1H NMR titration in DMSO-d6. For comparison, the anion binding property of macrocycle B2 and a control compound (CC) were also investigated. As shown in Fig. 1, upon addition of dicarboxylate (C72−), protons (NH, Hf) on the serine moiety and aromatic Hb of B2H showed continuous upfield shifts, which could be ascribed to deprotonation or proton exchange between serine carboxyl groups and the dicarboxylate guest. In stark contrast, the chemical shifts of aromatic proton Ha on the lower-rim of the macrocyclic unit firstly moved to upfield then sharply turned to downfield, giving a minimum turning point (Fig. 1). The whole binding process reached saturation upon addition of ca. 4.0 equiv. of dicarboxylates. As downfield movement of the lower-rim proton is indicative of anion–π binding [32], the complicated chemical shift changes of Ha therefore indicated mixed and competitive binding modes between B2H and dicarboxylates, i.e. the coexistence of proton exchange and anion‒π binding. Specifically, host B2H has the serine carboxyl groups to interact with dicarboxylates through proton exchanges, meanwhile dicarboxylate could also enter the macrocyclic cavity with each anionic head interacting with the electron-deficient V-shaped sub-cavity through anion–π interactions (for schematic illustration of the proposed binding equilibria, see Scheme 3).
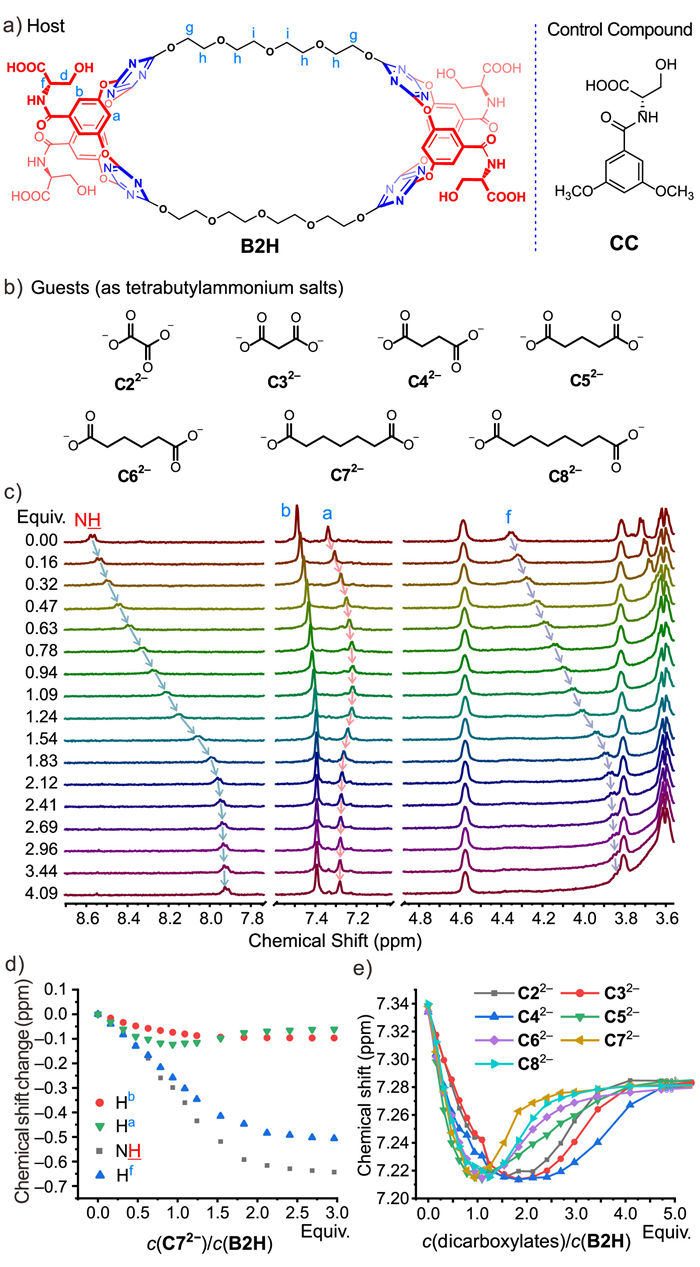
To further verify the hypothesis of different binding modes deduced from differentiated trends in chemical shift of Ha, several control titration experiments were conducted. When B2, the macrocycle with absence of serine carboxyl groups was applied as host and C62− as a representative guest, only a slight downfield-shift of NH proton was observed (0.35 ppm, Ka ~ 32.60 L/mol), indicating a simple weak hydrogen bonding between B2 and dicarboxylate and excluding anion–π binding (Fig. S15 in Supporting information). Such titration results highlight the dominant contribution of serine moieties in B2H–dicarboxylate interactions. Besides, we designed a control compound CC which only contains "lower-rim" Ha and a serine carboxyl group. Upon titration with dicarboxylate (C42−), only continuous upfield shifts of Ha were observed, which indicates a single deprotonation or proton exchange process (Fig. S16 in Supporting information). It is reasonable that the Ha alone could not show association ability with dicarboxylate because it is a very weak hydrogen bond donor. Only when Ha locates within the electron-deficient V-shaped cavity and when anion–π binding occurs it is forced to participate in the anion association through non-conventional hydrogen bond. On the other hand, we chose Cl− (Fig. S17, as tetrabutylammonium salt) to titrate host B2H. As Cl− is a weaker base in comparison with carboxylates, proton exchange is less possible in this case. Indeed, the titration spectra showed continuous downfield shift of Ha, corresponding to exclusive anion–π binding between B2H and Cl−.
Subsequently, we studied the binding properties of B2H towards dicarboxylates C22−–C82− (as tetrabutylammonium salt) with different chain lengths (Fig. 1e). 1H NMR titrations demonstrated that for all dicarboxylates the host exhibited consistent chemical shift changes (Figs. S7–S14 in Supporting information), indicating the similar proton exchange/anion–π binding modes (Scheme 3). However, the upfield-to-downfield turning points, and the titration isotherms of Ha were significantly dependent on the chain length of the dicarboxylates. For example, pimelate (C72−) caused a sharp turning point around 1.0 equiv. and the most significant downfield shifts after that, and was the fastest to reach the titration saturation among the dicarboxylate guests. Lengthening (C82−) or shortening (C62−–C22−) the alkyl chains of the dicarboxylates affected marginally the protonation process (before turning point), while resulting in less downfield chemical shift changes (anion–π binding) than C72− before saturation. The chemical shift changes could reflect the binding strength, which gave an order of C72− > C82− > C62−− > C52−− > C22− > C32− > C42−. The significant dependence of anion–π binding on length of dicarboxylates suggested that the size match between host and guest is important. The strongly bound dicarboxylate C72− probably represents the best-fit size corresponding to the separation of the two macrocyclic cavities and hence can make use of their cooperativity. Such assumption is also exemplified by the binding behaviors of C82−, C62− and C52−, which indicated that when the size of dianion derivates from C72−, weaker binding strength would be resulted. Strangely, for the short dicarboxylates C22−–C42−, instead of following the general size trend exhibited by other anions, a reverse order of C22− > C32− > C42− was observed. Besides, their upfield-to-downfield turning points are largely laggard, the minima of the chemical shift changes occurred until addition of ca. 2.0 equiv. anions. These unusual behaviors for C22−–C42− may suggest that the host B2H is capable of simultaneously binding two smaller anions to form 1:2 anion–π complexation. Notably, succinate (C42−) shows the weakest binding, probably due to its medium-length chain being neither long enough for 1:1 nor capable of squeezing a dimer for 1:2 anion–π complexation.
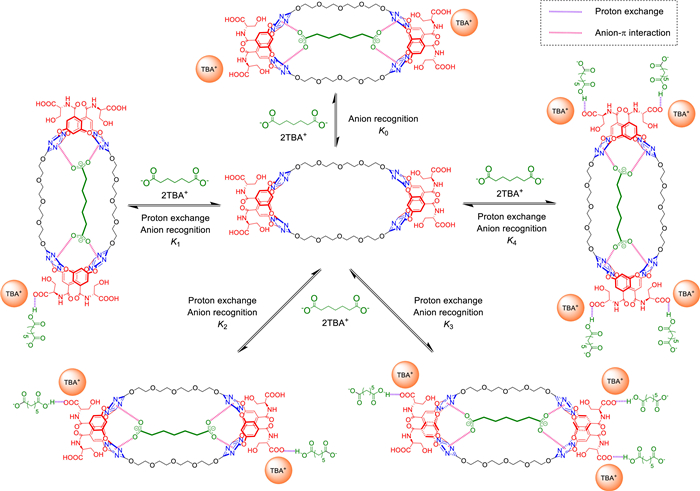
Based on the qualitative analyses above, it has been expected that further quantitative study could give us more information about binding stoichiometries and related association constants. Hopefully, by deep mining into the data of 1H NMR titrations, a non-linear fitting method has been harnessed to obtain these desired results. Since the host B2H could be regarded as a weak quaternary acid, the competitive balance of deprotonation or proton exchange and anion binding would continuously occur upon addition of basic anion titrants (for pKa of the anions see Table S4 and Fig. S19 in Supporting information). Considering all possible species in the solution and their relevant equilibrium constants (including their individual acidity constant Ka's and the mutual association constant K's between host and guest), combined with the expressions of material balance and charge balance, it is theoretically feasible to solve the equilibrium concentration distribution of each species during the titration process by means of a numerical iterative algorithm based on geometric programming method (Supporting information). Taking into account of the 1H NMR chemical shifts of different species, the functional relationship between the chemical shift curve of proton Ha and the unknown binding constants could be implicitly expressed. Finally, with the help of multi-variable nonlinear regression, a set of estimates of the unknown binding constants could be obtained.
In the specific calculation, we considered the different chemical shift curves of dicarboxylates (Fig. 2c) and OH− (Fig. S18 in Supporting information) as titrants because their diverse basicity could alter the best-fit range of the association constants of the anion with different stages of proton exchange of the host. In addition, the titration redistributed those proton-exchanged host species to emerge at their respective equivalents, the same way as the influence of association constants on the chemical shift curve (Fig. 2b). Therefore, the association constants of differently proton-exchanged host species could be independently and confidently fitted. For C72−, the association constants of the anion to the original host and its first two proton-exchanged species were K0 = (9.4 ± 3.4) × 103, K1 = (7.2 ± 1.4) × 103 and K2 = (3.4 ± 1.5) × 103 L/mol, respectively. It could be concluded that these bindings all showed relatively high affinity, but also followed an order of C72− > C82− > C62− > C52− (K) and gradually decreased along with the proton exchange of the host (Fig. 2d and Fig. S20 in Supporting information).

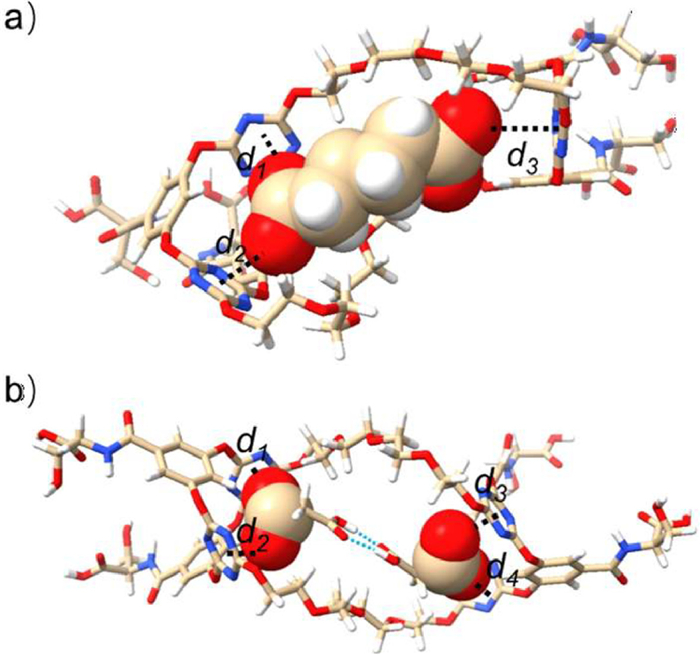
To visualize the proposed anion–π binding between the host and guest, we carried out geometry optimizations using M06–2X at the 6–311++G(d, p) level, taking 1:1 B2H—C62− and 1:2 B2H-[2C3H]2− complexes as typical examples. Fig. 3 showed the optimized conformations of these two complexes, where the single C62− anion and hydrogen bonded [2C3H]2− dimer are respectively encapsulated within the B2H cavity. The two sub-cavities interact synergistically with the included dicarboxylates. In both cases the short distances between triazine and the oxygen of carboxylate in the range of 2.8‒3.1 Å indicate multiple anion‒π interactions between host and guests. Besides, the glycol arms of the macrocycle might also assist the anion binding through van der Waals interaction with the alkyl chains of the dianions. Driven by the multiple noncovalent bonds, the host produces helical-like conformation after complexation (Fig. 3 and Fig. S21 in Supporting information). Optimization on B2H—C82− and B2H—C72− complexes gave similar results, with only the sub-cavity distances induced by the length of the guest dianion being different (Table S5 in Supporting information). The above results indicated that the macrocycle B2H, which bears serine moieties on the larger rim of each sub-macrocycle, showed enhanced anion binding ability and enabled competitive and different binding modes. Particularly, after deprotonation of the serine with dicarboxylates, the host in fact turns from charge-neutral to anionic nature which in principle is detrimental to further anion binding. However, our results clearly indicated the coexistence of anion–π complexation. It is difficult currently to thoroughly elucidate the whole process, we speculated that the counter cation (TBA+) might serve as a cationic bridge to facilitate the encapsulation of the dianion within the macrocyclic cavity (Fig. S13 in Supporting information).
In conclusion, we have demonstrated a straightforward one-pot strategy for the diversity-oriented construction of sophisticated ultracycles from rationally designed macrocyclic subunit precursors. The type and amount of the base were found to significantly influence the macrocyclization outcome. Existence of template anion could increase the total chemical yields and affect the distribution of the resulting macrocycles. The anion binding ability of B2H towards a series of dicarboxylates was investigated by means of 1H NMR titrations, which afforded interesting proton exchange/anion–π binding modes. Based on all possible species in the solution and their relevant equilibrium constants, and in combination of the material and charge balances, a numerical iterative algorithm was developed and applied to calculate the association constants of the anions (C52− to C82−) to the host B2H. It was shown that the binding affinity was relatively high with the association constants being up to 103 L/mol.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support from the National Natural Science Foundation of China (Nos. 22171271, 22022112) and Beijing National Laboratory for Molecular Sciences (No. BNLMS-CXXM-202002) is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
L.P. Yang, X. Wang, H. Yao, W. Jiang, Acc. Chem. Res. 53 (2020) 198–208. doi: 10.1021/acs.accounts.9b00415
J.M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley–VCH, 1995.
J.W. Steed, J.L. Atwood, Supramolecular Chemistry, 2nd ed., John Wiley & Sons, Ltd., 2009.
P. Neri, J.L. Sessler, M.X. Wang, Calixarenes and Beyond, Springer, 2016.
V. Prautzsch, S. Ibach, F. Vögtle, J. Incl. Phenom. Macro. 33 (1999) 427–457. doi: 10.1023/A:1017193104870
Z.Y. Zhang, C. Li, Acc. Chem. Res. 59 (2022) 916–929.
Y. Zhang, T. Wada, H. Sasabe, Chem. Commun. (1996) 621–622.
S.Q. Liu, D.X. Wang, Q.Y. Zheng, M.X. Wang, Chem. Commun. (2007) 3856–3858.
G. Gattuso, S. Menzer, S.A. Nepogodiev, J.F. Stoddart, D.J. Williams, Angew. Chem. Int. Ed. 36 (1997) 1451–1454. doi: 10.1002/anie.199714511
J. Jacob, K. Geβler, D. Hoffmann, et al., Angew. Chem. Int. Ed. 37 (1998) 606–609.
B. Li, B. Wang, X. Huang, et al., Angew. Chem. Int. Ed. 58 (2019) 3883–3889.
X.B. Hu, Z. Chen, L. Chen, et al., Chem. Commun. 48 (2012) 10999–11001. doi: 10.1039/c2cc36027f
X.J. Cheng, L.L. Liang, K. Chen, et al., Angew. Chem. Int. Ed. 52 (2013) 7252–7255. doi: 10.1002/anie.201210267
M. Iyoda, J. Syn. Org. Chem. Jpn. 70 (2012) 1157–1163. doi: 10.5059/yukigoseikyokaishi.70.1157
J. Gregoliński, K. Ślepokura, T. Paćkowski, et al., J. Org. Chem. 81 (2016) 5285–5294. doi: 10.1021/acs.joc.6b00531
C.F. Chen, L.G. Lu, Z.Q. Hu, X.X. Peng, Z.T. Huang, Tetrahedron 61 (2005) 3853–3858. doi: 10.1016/j.tet.2005.01.132
P. Lhoták, M. Kawaguchi, A. Ikeda, S. Shinkai, Tetrahedron 52 (1996) 12399–12408. doi: 10.1016/0040-4020(96)00738-7
N. Chopra, J.C. Sherman, Angew. Chem. Int. Ed. 36 (1997) 1727–1729. doi: 10.1002/anie.199717271
Z. Rodriguez-Docampo, E. Eugenieva-Ilieva, C. Reyheller, et al., Chem. Commun. 47 (2011) 9798–9800. doi: 10.1039/c1cc13451e
V. Valderrey, E.C. Escudero, Adán, P. Ballester, J. Am. Chem. Soc. 134 (2012) 10733–10736. doi: 10.1021/ja301900s
G.T. Hwang, B.H. Kim, Tetrahedron Lett. 41 (2000) 10055–10060. doi: 10.1016/S0040-4039(00)01818-9
S.Z. Li, K. Yang, H.B. Liu, et al., Tetrahedron Lett. 54 (2013) 5901–5906. doi: 10.1016/j.tetlet.2013.08.091
N.H. Evans, C.J. Serpell, K.E. Christensen, P.D. Beer, Eur. J. Inorg. Chem. 2012 (2012) 939–944. doi: 10.1002/ejic.201101257
Y. Singh, H.N. Hoang, B. Flanagan, D.P. Fairlie, Org. Lett. 8 (2006) 1053–1056. doi: 10.1021/ol052842e
F. Yang, Y. Ji, L. Zheng, H. Guo, J. Lin, Supramol. Chem. 18 (2006) 177–181. doi: 10.1080/10610270500398706
H.J. Hogben, J.K. Sprafke, M. Hoffmann, M. Pawlicki, H.L. Anderson, J. Am. Chem. Soc. 133 (2011) 20962–20969. doi: 10.1021/ja209254r
Q. Chen, L. Chen, C.Y. Wang, et al., Chem. Commun. 55 (2019) 13108–13111. doi: 10.1039/C9CC06692F
J. Luo, Y.F. Ao, Q.Q. Wang, D.X. Wang, Angew. Chem. Int. Ed. 57 (2018) 15827–15831. doi: 10.1002/anie.201810836
V. Martí. Centelles, M.D. Pandey, M.I. Burguete, S.V. Luis, Chem. Rev. 115 (2015) 8736–8834. doi: 10.1021/acs.chemrev.5b00056
P.S. Bols, H.L. Anderson, Acc. Chem. Res. 51 (2018) 2083–2092. doi: 10.1021/acs.accounts.8b00313
B.Y. Hou, Q.Y. Zheng, D.X. Wang, Z.T. Huang, M.X. Wang, Chem. Commun. (2008) 3864–3866.
R.B. Xu, Q.Q. Wang, Y.F. Ao, et al., Org. Lett. 19 (2017) 738–741. doi: 10.1021/acs.orglett.7b00070

Figure 1 (a) Structures of B2H and control compound CC. (b) Structure of dicarboxylates. (c) 1H NMR spectra of B2H upon titration with C72− (c(B2H) = 1 mmol/L, 298 K, 400 MHz, DMSO-d6). (d) Chemical shift changes of NH, Hb, Ha, Hf upon titration. (e) A plot of chemical shift of Ha versus titration equivalents c(dicarboxylates)/c(B2H).

Figure 2 (a) General scheme for the acquisition of association constants through nonlinear fitting method. (b) Equilibrium concentration distribution of B2H-related species upon titration with C62−. (c) Fitting results of the titration curves of dicarboxylate titrants C52− to C82−. (d) Fitting estimated association constants of dicarboxylates C52− to C82− with differently proton-exchanged B2H species (in logarithmic scale).

Figure 3 DFT optimized structures of (a) B2H—C62− and (b) B2H-[2C3H]2− complexes. The blue dotted lines represent hydrogen bonds.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

