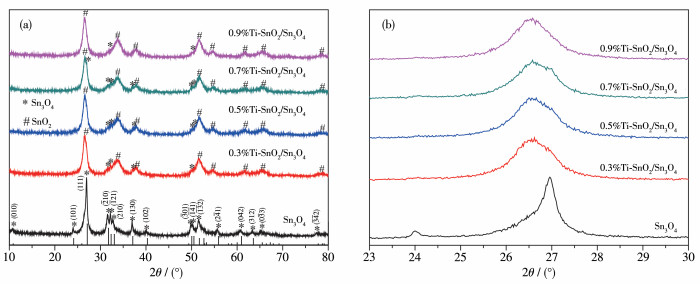
图 1.
Sn3O4和x%Ti-SnO2/Sn3O4样品的XRD图
Figure 1.
XRD patterns of Sn3O4 and x%Ti-SnO2/Sn3O4 samples

锡的单价氧化物只有SnO和SnO2,但混合价态的锡氧化物有多种,常见的有Sn2O3、Sn3O4、Sn5O6等。SnO是一种典型的p型半导体,能带宽度为2.7~3.2 eV,被广泛应用于气体传感器和锂离子电池正极材料[1-2]。SnO2是一种类似TiO2的宽禁带(3.6 eV)的n型半导体氧化物,具有耐热性、耐腐蚀性、制备成本低及对多种气体有良好响应等优点,常被用作气敏材料[3-4]。Sn3O4作为多价态锡氧化物的一种,是一种n型半导体材料,能带宽度比SnO2窄,且因其具有独特的结构、优异的光电化学性能,在光催化、锂离子电池及传感器领域的应用受到越来越多的关注[5]。
目前Sn3O4合成方法主要有水热法等,合成的形貌也是多种多样,有颗粒状、花球状、片状、空心球等[6-8]。例如,Berengue等[9]用水热法合成了颗粒状Sn3O4材料,并研究其在还原H2S制氢气中的应用,发现该方法制备的Sn3O4材料为大颗粒状,分散性不佳。Ma等[10]采用碳还原SnO2制备由一定的层状结构堆叠成的带状Sn3O4,并研究了该Sn3O4材料对氧气的气敏效应,但该制备方法的能耗较高。
半导体的电学、光学和催化性能与其晶体形态和微观结构密切相关。在各种形态结构的催化剂中,对于悬浮体系的光催化反应,三维纳/微结构的光催化性能优于低维平面几何结构[11],因为由单元结构堆叠而成的三维多尺度Sn3O4微米球通常拥有较大的比表面积,并且可引起光多重反射,有利于增强对光的捕获,同时在光催化反应中能够提供更多的吸附位点[12-14]。
单一相态的Sn3O4虽然具有明显的光催化性能,但其较低的光生电子-空穴分离效率,限制了其光催化性能的进一步提升[15-16]。对于单一半导体光生电子-空穴分离效率低的问题,常通过贵金属沉积、离子掺杂、构建异质相结等方法提升载流子的分离效率[17-19]。例如,Li等[20]通过简单的两步水热法制备Z-型Sn3O4/g-C3N4异质结构,提高了载流子的分离效率,这在很大程度上提高了其在水中对盐酸四环素的去除效率。另有报道发现Sn3O4与还原氧化石墨烯耦合后,能提高其析氢的稳定性[21]。除此之外,贵金属沉积、元素掺杂也是比较理想的提高光催化性能的手段[22-25]。因此可以在掺杂元素的同时实现构建异质相结,利用掺杂和异质相结的协同作用提高Sn3O4光催化性能。我们在水热条件下,通过Ti4+掺杂可以构建具有三维SnO2/Sn3O4异质相结的微米球光催化剂。研究发现,Ti4+掺杂和异质相结的生成,可以有效调节Sn3O4的带隙、增强光捕获能力、提高载流子分离效率。制备的Ti-SnO2/Sn3O4在去除Cr(Ⅵ) 和有机污染物中表现出优越的光催化性能。
称取1.128 3 g SnCl2·2H2O溶解在60 mL去离子水中,在搅拌过程中加入适量柠檬酸钠(Na3C6H5O7· 2H2O), 混合均匀,搅拌10 min,用1 mol·L-1 NaOH调节溶液pH=5.5,剧烈搅拌下加入n μL(n=12.9、21.5、30.2、38.8) TiCl3溶液,搅拌1 h后转移至含有聚四氟乙烯衬里的不锈钢高压反应釜,在180 ℃反应12 h后,冷却至室温,将沉淀离心,用无水乙醇和去离子水交替洗涤3次,最后在60 ℃烘箱中干燥12 h,获得x%Ti-SnO2/Sn3O4(x%为Ti与Sn的物质的量之比)。Sn3O4的制备过程与x%Ti-SnO2/Sn3O4相似,只是不添加TiCl3。
采用Ultima Ⅳ型X射线粉末衍射仪(XRD,Cu Kα,λ=0.154 06 nm,扫描速率5 (°)·min-1,工作电压40 kV,电流为40 mA)分析样品结构。采用FEI Talos F200S Super-X型透射电镜(TEM)和高分辨率透射电镜(HRTEM,加速电压200 kV)和HITACHI公司的Regulus8220型扫描电子显微镜(SEM,加速电压0.2~30 kV,着陆电压0.05~30 kV)对催化剂样品进行微观结构和形貌表征。采用美国麦克公司的ASAP2020型物理吸附仪器测定样品的比表面积及孔容、孔径(脱气温度90 ℃,脱气时间1 h)。使用Nicolet 6700 FT-IR光谱仪获得傅里叶变换红外光谱。使用DXR2型拉曼光谱仪测量样品的拉曼光谱。元素的化学价在Thermo Scientific K-Alpha型X射线光电子能谱仪(XPS)上进行分析。采用日本岛津UV-2550紫外可见分光光度计对样品进行紫外可见漫反射光谱分析(BaSO4标准品校准)。使用日立F -4600荧光光谱仪研究样品的光致发光谱(PL)。电化学测试在电化学工作站(CHI660E A15459)上进行,电解液为0.1 mol·L-1 Na2SO4,参比电极为Ag/ AgCl,辅助电极为Pt。工作电极的制备:称取10 mg样品加入200 μL乙醇溶液,超声分散30 min后,将样品均匀滴加在导电玻璃上(滴加面积:1 cm×1 cm),利用指甲油封边,用紫外灯烘干。
以Cr(Ⅵ)(20 mg·L-1)和偶氮染料(10 mg·L-1甲基橙(MO)和10 mg·L-1酸性橙Ⅱ(AO-Ⅱ))为模拟污染物,光源为400 W金卤灯(波长范围:350~2 500 nm),反应过程中通循环冷却水以保持反应体系在室温下恒定。称取催化剂样品30 mg分散于50 mL模拟污染物溶液中,在黑暗搅拌30 min达到物理吸附- 脱附平衡。在反应过程中取样3 mL,离心去除催化剂粉末,取上清液,用紫外-可见分光光度计(UV-2550)分析模拟污染物浓度的变化。Cr(Ⅵ)的浓度采用1,5-二苯羰酰肼显色法进行测定[13-14]。
从图 1可以清晰地看到,纯Sn3O4在2θ=10.8°、24.1°、27.1°、31.7°、32.3°、33°和37.1°处的衍射峰可归为(010)、(101)、(111)、(210)、(121)、(210)和(130)晶面衍射峰,对应三斜晶系的Sn3O4(PDF No.16.0737),衍射峰尖锐且清晰,表明样品的结晶度良好。掺入Ti4+后样品的主衍射峰发生变化,由原来的2θ=27.1°变为2θ=26.5°,对比衍射峰位可知生成了新的SnO2晶相(PDF No.77-0450)且为四方晶系,结晶度良好,但是仍有Sn3O4的衍射峰。由于Ti4+离子半径为53 pm,比Sn4+离子半径(69 pm)小[26]。因此,Ti4+可以在Sn3O4和SnO2的晶格中取代Sn4+,形成替代掺杂,且对晶格影响较小。XRD图表明Ti4+掺杂后一部分Sn4+与O结合直接生成新晶相SnO2,形成了SnO2/ Sn3O4复合物,并非单一的Sn3O4物质。
锡氧化物的生成受到Na3C6H5O7·2H2O的添加比例、pH、水热温度及时间的影响,在水热合成中,(C6H5O7)3-作为Sn配位剂,而pH值会影响配位剂的配位数[27]。因此,在相同水热温度及时长条件下,(C6H5O7)3-/Sn2+物质的量之比(n(C6H5O7)3-/nSn2+)和pH值决定了水热合成产物的相。研究表明[28],在0.75 < n(C6H5O7)3-/nSn2+ < 5.0、5.3 < pH < 6.8条件下水热生成纯Sn3O4相,改变n(C6H5O7)3-/nSn2+或pH值会生成SnO2相或SnO相。加入TiCl3后会改变溶液pH值,使得pH值处于临界状态,导致生成一部分SnO2覆盖在Sn3O4表面[29]。
Sn3O4和x%Ti-SnO2/Sn3O4的SEM图如图 2所示,Sn3O4是由纳米片自组装的花瓣状微球,该特殊形态具有多孔结构、大比表面积等特点。掺入Ti4+后,样品的形貌发生了显著变化,在纳米片上包裹了大量的纳米微球颗粒,且随着Ti4+掺杂量的增加,纳米片上包裹的颗粒越来越多,同时样品的形貌逐渐从规则的花球状趋向逐渐分散的团聚状。

为了揭示Ti4+掺杂引起了形貌的变化及证实SnO2的生成,对其进行了TEM分析。由图 3a1可知,分散覆盖在纳米片上的微球颗粒直径为~80 nm,粒径均匀。在典型纳米片上进行HRTEM分析,如图 3a2所示,0.359和0.283 nm的晶格间距分别对应Sn3O4的(101)和(210)晶面。同样对纳米片上覆盖的纳米球颗粒进行HRTEM分析可知,表面球形颗粒的晶格间距为0.335、0.239和0.265 nm(图 3a3),分别对应SnO2的(110)、(200)和(101)晶面,表明纳米片上覆盖的微球颗粒主要为SnO2晶相。图 3b的元素成像图表明Ti元素均匀掺杂在锡氧化物中。

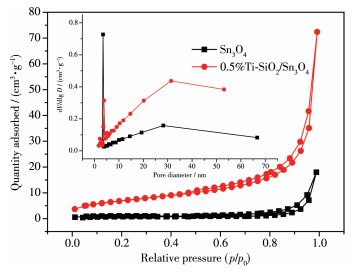
采用N2吸附-脱附等温线对合成样品的孔径分布(插入图)和比表面积进行研究。从图 4中可以看出,样品Sn3O4和0.5%Ti-SnO2/Sn3O4的N2吸附-脱附等温线均属于典型Ⅳ型,表明样品中有大量微孔存在[30],而样品回滞环均属于H3类型,这表明样品是由片状结构堆叠而成的狭缝孔[31]。图中插图为样品Sn3O4和0.5%Ti-SnO2/Sn3O4的孔径分布图,从图中可以看出样品的孔径大致分布在2~70 nm内,表明样品中存在介孔和大孔。样品Sn3O4的孔大概率分布在4 nm左右,而0.5%Ti-SnO2/Sn3O4的孔大部分分布在30 nm左右。从表 1中可以看出,Ti4+掺杂后样品比表面积均有所增大,且0.5%Ti-SnO2/Sn3O4的比表面积最大。
 下载:
导出CSV
下载:
导出CSV
| Sample | Specific surface area / (m2·g-1) | Pore volume / (cm3·g-1) | Pore size / nm |
| Sn3O4 | 3 | 0.027 8 | 11.25 |
| 0.3%Ti-SnO2/Sn3O4 | 22 | 0.116 9 | 20.54 |
| 0.5%Ti-SnO2/Sn3O4 | 25 | 0.112 0 | 17.32 |
| 0.7%Ti-SnO2/Sn3O4 | 20 | 0.099 7 | 19.07 |
| 0.9%Ti-SnO2/Sn3O4 | 24 | 0.137 5 | 23.59 |
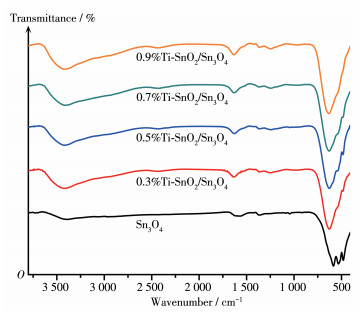
利用FT-IR对样品的表面化学键性质进行研究。由图 5可知,在3 420和1 630 cm-1处的吸收峰分别为—OH伸缩振动和吸附水的变形振动峰[32]。随着Ti4+掺杂量的增加,吸收峰强度有所增大,说明样品表面具有较丰富的—OH,这有利于提高光催化活性[33]。波数为1 366 cm-1处的吸收峰为样品吸附CO2的C=O伸缩振动峰[34]。另外,在400~600 cm-1处的吸收带均可归属于Sn—O键振动带[35]。掺杂Ti4+后位于483和533 cm-1处的Sn—O振动峰明显减弱,而位于584 cm-1处的不对称Sn—O—Sn振动峰明显,也证明掺杂Ti4+对Sn—O键产生了影响。
通过XPS分析0.5%Ti-SnO2/Sn3O4样品的化学组成和元素价态,并将0.5%Ti-SnO2/Sn3O4和Sn3O4作对比分析Ti4+掺杂导致的化学环境的变化。在图 6a的全谱图中可以看到SnO2/0.5%Ti-Sn3O4和Sn3O4样品中都含有Sn、O和C元素的能谱峰,但由于Ti元素掺杂含量较低,在低分辨全谱中未发现Ti元素能谱峰。对各元素进行高分辨XPS分析,使用C1s标准碳峰校准。在图 6b可以看出,0.5%Ti-SnO2/Sn3O4和Sn3O4样品中的O1s分别位于530.54和530.45 eV,由于Ti4+的掺杂导致O1s向高结合能发生略微偏移。0.5%Ti-SnO2/Sn3O4位于530.54 eV处的结合能可以拟合出4个峰,位于530.50、531.04和529.97 eV处的结合能分别对应O—Sn2+和O—Sn4+以及Ti—O[36],位于532.03 eV处归属于吸附水中的O—H键。同样的偏移现象在图 6c的Sn3d谱图中也可以观察到。在经过Ti4+掺杂后,Sn3d的2个峰(494.92和486.48 eV)向高结合能偏移0.2 eV(495.12和486.67 eV)。将Sn3d中的这2个峰进行拟合可以得到2组峰。在Sn3O4中,495.00和486.58 eV分别对应Sn4+的Sn3d3/2和Sn3d5/2,而结合能位于494.75和486.33 eV处的2个峰则分别对应Sn2+的Sn3d3/2和Sn3d5/2。在0.5%Ti-SnO2/Sn3O4中,结合能495.19和486.72 eV,分别对应于Sn4+的Sn3d3/2和Sn3d5/2,而结合能位于494.95和486.50 eV处的2个峰则分别对应Sn2+的Sn3d3/2和Sn3d5/2。这是因为Ti4+的离子半径较锡氧化物中Sn4+的离子半径更小,因此对外层电子的束缚能力更强,因此替换成键后导致结合能向高处偏移。在Ti2p的高分辨谱图(图 6d)中,结合能位于458.58和464.32 eV处的2个峰分别对应于Ti2p3/2和Ti2p1/2,表明样品中Ti为+4价。
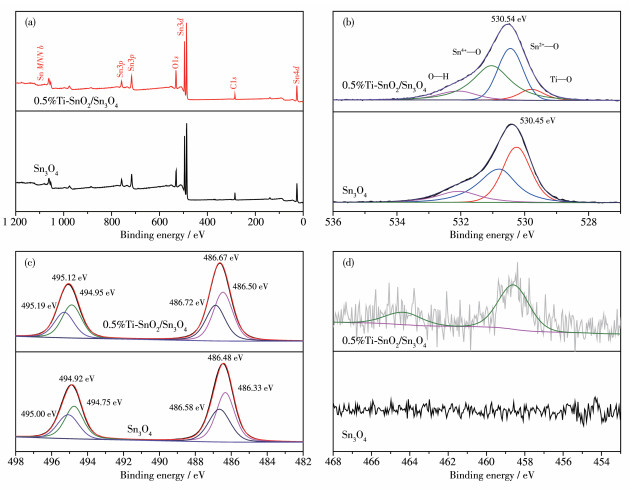
(a) Survey spectrum; (b) O1s; (c) Sn3d; (d) Ti2p
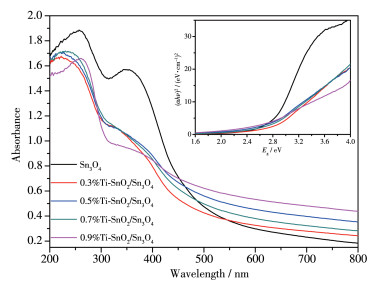
图 7为样品的UV-Vis DRS光谱图。从图中可以看出样品在紫外和可见光区域对光均有吸收。Ti4+掺杂后的样品较纯样在紫外区吸收强度有所减弱。另外,Ti4+掺杂使样品在可见光区的吸收边缘发生一定的红移。根据Tauc曲线估算出图中Sn3O4和x%Ti-SnO2/Sn3O4的间接带隙(Eg)值。Tauc曲线由紫外可见光谱中的(αhν)2和hν进行转换,其中α、h和ν分别为吸收系数、普朗克常数和光频率[37]。样品Eg值见表 2。Sn3O4的Eg值为2.77 eV,而0.5%Ti-SnO2/ Sn3O4的Eg值为2.59 eV。Ti4+的掺入使得Sn3O4带隙发生变化,且随着Ti4+掺杂量增加,Eg值逐渐减小。
 下载:
导出CSV
下载:
导出CSV
| Sample | Eg / eV |
| Sn3O4 | 2.77 |
| 0.3%Ti-SnO2/Sn3O4 | 2.76 |
| 0.5%Ti-SnO2/Sn3O4 | 2.59 |
| 0.7%Ti-SnO2/Sn3O4 | 2.52 |
| 0.9%Ti-SnO2/Sn3O4 | 2.42 |
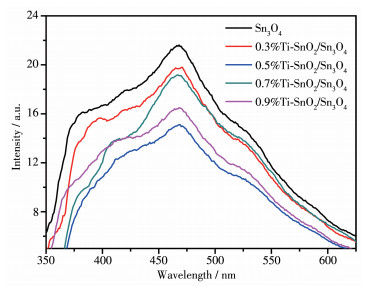
PL谱图可用于研究半导体载流子的转移复合情况,光致发光光谱的发射峰强度越低,表明载流子复合效率越低。从图 8可以清楚地看出,与Sn3O4相比,x%Ti-SnO2/Sn3O4的PL强度明显降低,进一步证明了Ti4+掺杂可以抑制光诱导电子-空穴对的重组。适当增加掺杂含量可以有效地减弱光致发光强度,其中0.5%Ti-SnO2/Sn3O4的光致发光强度最低。全面分析表明,Ti4+掺杂引起SnO2晶相的生成,形成SnO2/Sn3O4异质界面,在异质界面之间存在电势差,这样可以有效地转移光生载流子,提高其分离效率。
在瞬态光电流响应分析中,光电流越强,光生载流子分离效率越高。从图 9a中可以清楚地看到,在光照条件下,Sn3O4和x%Ti-SnO2/Sn3O4均有快速的光电流响应。值得注意的是,x%Ti-SnO2/Sn3O4的光电流密度明显强于Sn3O4,这是由于Ti4+掺杂增强了光生载流子的产生和分离,而0.5%Ti-SnO2/Sn3O4表现出最强的瞬态光电流。
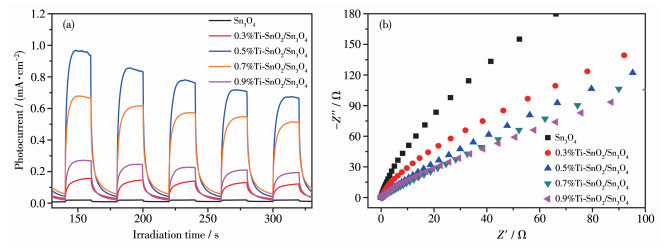
电化学阻抗谱(EIS)是揭示电子在电极上的转移和分离效率的另一种有效的电化学方法。研究认为,小的EIS弧半径表明工作电极与电解质溶液之间的电荷转移电阻小,即工作电极具有更有效的光生电荷转移能力。如图 9b所示,与Sn3O4相比,x%Ti-SnO2/Sn3O4的EIS弧半径随着Ti4+含量的增加而减小,进一步说明Ti4+掺杂后增强了光生电子空穴对的分离和转移。
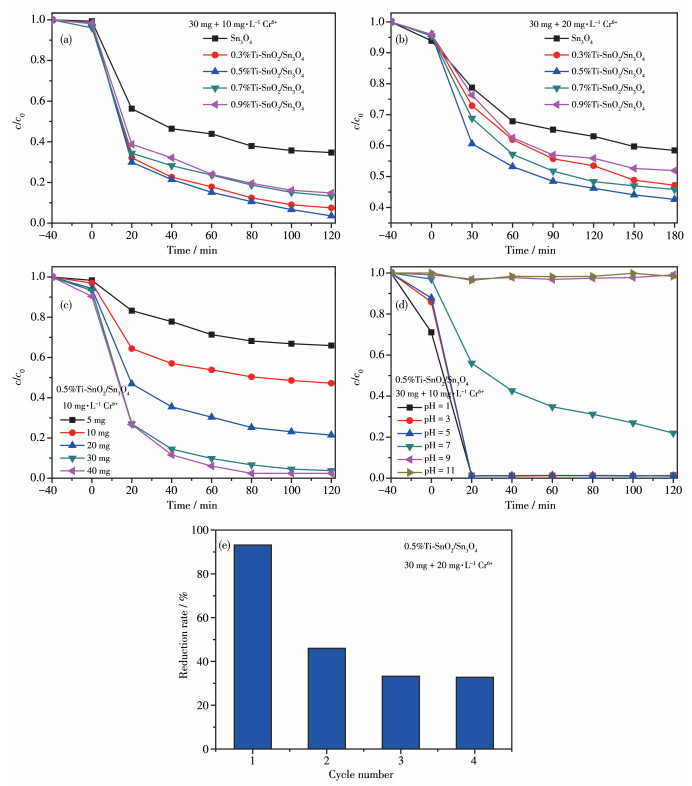
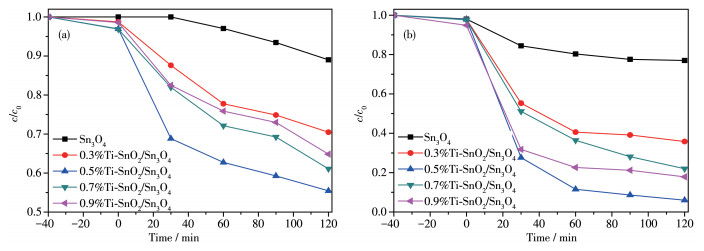
首先通过还原Cr(Ⅵ)来评价样品的光催化性能。从图 10a中可以明显观察到纯Sn3O4样品本身具有一定的光催化还原效果,在120 min后还原率达到62%,而x%Ti-SnO2/Sn3O4均表现出较纯样更高的还原性。其中0.5%Ti-SnO2/Sn3O4在120 min后对Cr(Ⅵ) 还原率达到96%。将Cr(Ⅵ)的浓度从10 mg·L-1增加到20 mg·L-1继续考察污染物浓度对还原效果的影响(图 10b)可知,Cr(Ⅵ)浓度的增加使催化剂的还原率有所下降,但掺杂样仍较纯样表现出较高的还原率,其中0.5%Ti-SnO2/Sn3O4对Cr(Ⅵ)还原率达到56%。图 10c为0.5%Ti-SnO2/Sn3O4的使用量对10 mg·L-1 Cr6+的还原性能的影响,由图可知,最佳催化剂使用量为30 mg。图 10d为30 mg的0.5%Ti-SnO2/Sn3O4在不同pH值下对10 mg·L-1 Cr6+的还原性能比较。发现溶液的酸碱性对还原活性有重大影响,在碱性条件下几乎不对Cr6+进行还原,而在中性及酸性条件下还原率增加,尤其是强酸性条件下,20 min即可去除Cr6+,还原率达99%,而实验室配制的Cr6+溶液pH值约为6.4。图 10e为0.5%Ti-SnO2/Sn3O4在20 mg·L-1 Cr6+中的稳定性测试,循环实验发现,第2次循环时,还原效果只有第1次的一半,第3次及第4次还原效果相差不大,趋于稳定,但整体还原效果只有第1次的1/3,因此,样品循环使用性能一般。
进一步通过降解染料类有机污染物来评价样品的光催化剂活性。图 11a为30 mg样品在400 W金卤灯照射下对MO的降解效率。可以发现,Sn3O4本身对MO的降解活性不大,光照120 min对MO降解率仅22%,而Ti4+掺杂后样品降解率显著提升,最佳样品0.5%Ti-SnO2/Sn3O4在120 min内对MO的降解率达到94%。同样,对AO-Ⅱ降解表现出相似的活性规律,但在相同时间内,催化剂对AO-Ⅱ的降解活性不如对MO的活性大,最佳样品0.5%Ti-SnO2/ Sn3O4的降解率达45%。
通过UV-Vis DRS结果得到Sn3O4的带隙为2.77 eV,SnO2的带隙为3.36 eV[38]。根据电负性原理计算两者的导带电势(ECB)和价带电势(EVB),公式如下:
|
|
其中,X为半导体的绝对电负性,Ee为氢标准下的自由电子能量,通常为4.5 eV,Eg为半导体的带隙。Sn3O4的绝对电负性为5.93,SnO2的绝对电负性为6.25[39]。因此可以得出Sn3O4的ECB和EVB分别为0.045和2.815 eV;SnO2的ECB和EVB分别为0.07和3.43 eV。掺杂Ti4+之后的Sn3O4带隙略有缩小。提出的Ti-SnO2/Sn3O4光催化活性提高机理如图 12所示。在全光谱光辐射下,SnO2和Sn3O4都能够被激发,在能级差形成的界面电场推动下,Ti-Sn3O4激发的电子能够迁移至SnO2的导带上,进而能够转移至Ti-SnO2/Sn3O4的表面与Cr(Ⅵ)发生还原反应。与此同时,SnO2价带上的空穴可转移至Ti-Sn3O4的价带上,该电子和空穴的转移减小了其内部复合,提高了载流子浓度和利用效率,进而提升了光催化活性。
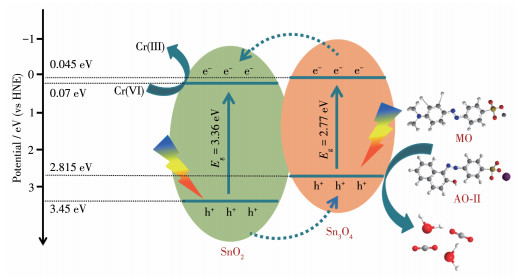
通过水热法制备了花球状Sn3O4及x%Ti-SnO2/Sn3O4微米球。研究发现,Ti4+掺杂引起光催化活性增强的主要原因如下:首先,金属离子Ti4+进入Sn3O4晶格中替代Sn4+,形成替代掺杂,由于离子半径的不同,造成晶格发生一定程度的畸变,引起晶格缺陷,形成更多的氧空位;其次,SnO2/Sn3O4异质结的生成进一步提升了光生电子与空穴的分离效率,同时增大了催化剂比表面积和可见光区的光吸收能力。因此在异质结和掺杂共同作用下,提高了Sn3O4光催化氧化还原性能。
Guo Q, Wang G X, Kumar A, Pandey R. Nanotechnology, 2017, 28(47): 475708 doi: 10.1088/1361-6528/aa92ab
Miller S A, Gorai P, Aydemir U, Mason T O, Stevanović V, Toberer E S, Snyder G J. J. Mater. Chem. C, 2017, 5: 8854-8861 doi: 10.1039/C7TC01623A
Wei Y J, Zheng H, Hu S S, Pu S Z, Peng H Y, Li L, Sheng H P, Zhou S Y, Wang J B, Jia S F. CrystEngComm, 2018, 20: 7114-7119 doi: 10.1039/C8CE01507D
Matussin S, Harunsani M H, Tan A L, Khan M M. ACS Sustainable Chem. Eng. , 2020, 8(8): 3040-3054 doi: 10.1021/acssuschemeng.9b06398
Song H, Son S, Kim S K, Jung G Y. Nano Res. , 2015, 8(11): 3553-3561 doi: 10.1007/s12274-015-0855-2
Liu J Y, Wang C, Yang Q Y, Gao Y, Zhou X, Liang X S, Sun P, Lu G Y. Sens. Actuators B Chem. , 2016, 224: 128-133 doi: 10.1016/j.snb.2015.09.089
Manikandan M, Tanabe T, Li P, Ueda S, V. Ramesh G, Kodiyath R, Wang J, Hara T, Dakshanamoorthy A, Ishihara S, Ariga K, Ye J, Umezawa N, Abe H. ACS Appl. Mater. Interfaces, 2014, 6(6): 3790-3793 doi: 10.1021/am500157u
Xu W, Li M, Chen X B, Zhao J H, Tan R Q, Li R, Li J, Song W J. Mater. Lett. , 2014, 120: 140-142 doi: 10.1016/j.matlet.2014.01.075
Berengue O M, Simon R A, Chiquito A J, Dalmaschio C J, Leite E R, Guerreiro H A, Guimarães F E G. J. Appl. Phys. , 2010, 107(3): 033717-033717 doi: 10.1063/1.3294613
Ma X H, Shen J L, Hu D X, Sun L, Chen Y, Liu M, Li C N, Ruan S P. J. Alloys Compd. , 2017, 726: 1092-1100 doi: 10.1016/j.jallcom.2017.08.079
Ji L J, Zhang Y H, Miao S Y, Gong M D, Liu X. Carbon, 2017, 125: 544-550 doi: 10.1016/j.carbon.2017.09.094
Xia W W, Qian H Y, Zeng X, Dong J, Wang J, Xu Q. J. Phys. Chem. C, 2017, 121(35): 19036-19043 doi: 10.1021/acs.jpcc.7b05520
Zeng D B, Yu C L, Fan Q Z, Zeng J L, Wei L F, Li Z S, Yang K, Ji H B. Chem. Eng. J. , 2020, 391: 123607 doi: 10.1016/j.cej.2019.123607
Yu C L, Zeng D B, Fan Q Z, Yang K, Zeng J L, Wei L F, Yi J H, Ji H B. Environ. Sci. : Nano, 2020, 7: 286-303 doi: 10.1039/C9EN00899C
Niu P, Qiao M, Li Y F, Huang L, Zhai T Y. Nano Energy, 2018, 44: 73-81 doi: 10.1016/j.nanoen.2017.11.059
Chen X F, Huang Y, Zhang K C, Feng X, Wang M. Electrochim. Acta, 2018, 259: 131-142 doi: 10.1016/j.electacta.2017.10.180
翟春阳, 孙明娟, 杜玉扣, 朱明山. 无机材料学报, 2017, 32(9): 897-903 https://www.cnki.com.cn/Article/CJFDTOTAL-ZGZS201903001.htmZHAI C Y, SUN M J, DU Y K, ZHU M S. J. Inorg. Mater. , 2017, 32(9): 897-903 https://www.cnki.com.cn/Article/CJFDTOTAL-ZGZS201903001.htm
Sun M, He Z L, Yuan C, Wang X D, Zhai C Y, Zhu M S. Ind. Eng. Chem. Res. , 2020, 60(1): 762-770
Wang X D, Gao H F, Zhai C Y, He Z L, Yuan C, Zhu M S. Ind. Eng. Chem. Res. , 2020, 59(43): 19252-19259 doi: 10.1021/acs.iecr.0c03436
Li C M, Yu S Y, Dong H J, Liu C B, Wu H J, Che H N, Chen G. Appl. Catal. B, 2018, 238: 284-293 doi: 10.1016/j.apcatb.2018.07.049
Yu X, Zhao Z H, Sun D H, Ren N, Yu J G, Yang R Q, Liu H. Appl. Catal. B, 2018, 227: 470-476 doi: 10.1016/j.apcatb.2018.01.055
Wang W, Moses O T, Shao Z. Prog. Mater Sci. , 2018, 92: 33-63 doi: 10.1016/j.pmatsci.2017.09.002
陈范云, 张萌迪, 马小帅, 李家德, 余长林. 无机化学学报, 2019, 35(6): 1034-1040 doi: 10.11862/CJIC.2019.115CHEN F Y, ZHANG M D, MA X S, LI J D, YU C L. Chinese J. Inorg. Chem. , 2019, 35(6): 1034-1040 doi: 10.11862/CJIC.2019.115
马小帅, 陈范云, 余长林, 杨凯, 黄微雅, 李韶雨. 无机化学学报, 2020, 36(2): 217-225 doi: 10.11862/CJIC.2020.040MA X S, CHEN F Y, YU C L, YANG K, HUANG W Y, LI S Y. Chi-nese J. Inorg. Chem. , 2020, 36(2): 217-225 doi: 10.11862/CJIC.2020.040
何洪波, 张梦凡, 刘珍, 樊启哲, 杨凯, 余长林. 无机化学学报, 2020, 36(8): 1413-1420 doi: 10.11862/CJIC.2020.177HE H B, ZHANG M F, LIU Z, FAN Q Z, YANG K, YU C L. Chinese J. Inorg. Chem. , 2020, 36(8): 1413-1420 doi: 10.11862/CJIC.2020.177
Yao X J, Tang C J, Ji Z Y, Dai Y, Cao Y, Gao F, Dong L, Chen Y. Catal. Sci. Technol, 2013, 3: 688-698 doi: 10.1039/C2CY20610B
Han C F, Liu Q, Ivey D G. Electrochim. Acta, 2008, 53(28): 8332-8340 doi: 10.1016/j.electacta.2008.06.037
Tanabe T, Hashimoto M, Mibu K, Tanikawa T, Gunji T, Kaneko S, Abe H, Miyauchi M, Matsumoto F. J. Nanosci. Nanotechnol. , 2017, 17(5): 3454-3459 doi: 10.1166/jnn.2017.13060
韦志仁, 罗小平, 付三玲, 李哲, 胡志鹏, 高平, 王伟伟, 董国义. 人工晶体学报, 2007, 36(6): 1301-1304 doi: 10.3969/j.issn.1000-985X.2007.06.021WEI Z R, LUO X P, FU S L, LI Z, HU Z P, GAO P, WANG W W, DONG G Y. Journal of Synthetic Crystals, 2007, 36(6): 1301-1304 doi: 10.3969/j.issn.1000-985X.2007.06.021
Xiang Q J, Yu J G, Mietek J. J. Phys. Chem. C, 2011, 115(15): 7355-7363 doi: 10.1021/jp200953k
Liu R Y, Wu Z, Tian J, Yu C L, Li S Y, Yang K, Liu X Q, Liu M C, New J. Chem. , 2018, 42: 137-149
刘守新, 刘鸿. 光催化及光电催化基础与应用. 北京: 化学工业出版社, 2005: 51-58LIU S X, LIU H. Basic and Application of Photocatalysis and Photocatalysis. Beijing: Chemical Industry Press, 2005: 51-58
赵杰, 赵经贵, 高山, 霍丽华. 光散射学报, 2004, 3: 234-236 doi: 10.3969/j.issn.1004-5929.2004.03.009ZHAO J, ZHAO J G, GAO S, HUO L H. The Journal of Light Scattering, 2004, 3: 234-236 doi: 10.3969/j.issn.1004-5929.2004.03.009
Saranya M, Ramachandran R, Samuel E J J, Jeong S K, Grace A N. Powder Technol. , 2015, 279: 209-220 doi: 10.1016/j.powtec.2015.03.041
Li C M, Yu S Y, Dong H J, Liu H J, Che H N, Chen G. Appl. Catal. B, 2018, 238: 284-293 doi: 10.1016/j.apcatb.2018.07.049
Moses P R, Larry M W, John C L, Flnklea H O, Lenhard J R, Murray R W. Anal. Chem. , 1978, 50(4): 576-585 doi: 10.1021/ac50026a010
Cho S, Ahn C, Park J, Jeon S. Nanoscale, 2018, 10: 9747-9751 doi: 10.1039/C8NR02330A
Kumar M R, Murugadoss G, Venkatesh N, Sakthivel P. ChemistrySelect, 2020, 5(23): 6946-6953 doi: 10.1002/slct.202001227
Pearson, Ralph G. Inorg. Chem. , 1988, 27(4): 734-740 doi: 10.1021/ic00277a030

图 1 Sn3O4和x%Ti-SnO2/Sn3O4样品的XRD图
Figure 1 XRD patterns of Sn3O4 and x%Ti-SnO2/Sn3O4 samples

图 2 Sn3O4与x%Ti-SnO2/Sn3O4样品SEM图: (a1、a2) Sn3O4; (b) 0.3%Ti-SnO2/Sn3O4; (c) 0.5%Ti-SnO2/Sn3O4; (d) 0.7%Ti-SnO2/Sn3O4; (e) 0.9%Ti-SnO2/Sn3O4
Figure 2 SEM images of Sn3O4 and x%Ti-SnO2/Sn3O4 samples: (a1, a2) Sn3O4; (b) 0.3%Ti-SnO2/Sn3O4; (c) 0.5%Ti-SnO2/Sn3O4; (d) 0.7%Ti-SnO2/Sn3O4; (e) 0.9%Ti-SnO2/Sn3O4

图 3 0.5%Ti-SnO2/Sn3O4样品的(a1) TEM图、(a2、a3) HRTEM图和(b1~b4)相应的元素成像图
Figure 3 (a1) TEM image, (a2, a3) HRTEM images and (b1~b4) corresponding element mappings of 0.5%Ti-SnO2/Sn3O4 sample

图 4 Sn3O4和0.5%Ti-SnO2/Sn3O4的N2吸附-脱附等温线及孔径分布曲线(插图)
Figure 4 N2 adsorption-desorption isotherms and pore size distribution curves of Sn3O4 and 0.5%Ti-SnO2/Sn3O4

图 5 Sn3O4与x%Ti-SnO2/Sn3O4样品的FT-IR谱图
Figure 5 FT-IR spectra of Sn3O4 and x%Ti-SnO2/Sn3O4 samples

图 6 0.5%Ti-SnO2/Sn3O4和Sn3O4样品的XPS谱图
Figure 6 XPS spectra of 0.5%Ti-SnO2/Sn3O4 and Sn3O4 sample
(a) Survey spectrum; (b) O1s; (c) Sn3d; (d) Ti2p

图 7 样品的UV-Vis DRS谱图和带隙(αhν)2-Eg谱图(插图)
Figure 7 UV-Vis DRS spectra and band gap (αhν)2-Eg spectra of the samples

图 8 Sn3O4和x%Ti-SnO2/Sn3O4样品的PL谱图
Figure 8 PL spectra of Sn3O4 and x%Ti-SnO2/Sn3O4 samples

图 9 样品的电化学测试: (a) 瞬态光电流密度响应图; (b) Nyquist图
Figure 9 Electrochemical test of the samples: (a) transient photocurrent density response of samples; (b) Nyquist plots

图 10 样品还原Cr6+的活性图: (a) 催化剂样品去除10 mg·L-1 Cr6+的活性图; (b) 催化剂样品去除20 mg·L-1 Cr6+的活性图; (c) 不同含量的0.5%Ti-SnO2/Sn3O4还原10 mg·L-1 Cr6+的活性图; (d) 30 mg 0.5%Ti- SnO2/Sn3O4还原不同pH值的10 mg·L-1 Cr6+的活性图; (e) 0.5%Ti-SnO2/Sn3O4的循环实验
Figure 10 Activity diagram of samples for reduction of Cr6+: (a) activity diagram of the catalyst sample for the removal of 10 mg·L-1 Cr6+; (b) activity diagram of the catalyst sample to remove 20 mg·L-1 Cr6+; (c) activity diagram of 10 mg·L-1 Cr6+ reduction by 0.5%Ti-SnO 2/Sn3O4 with different contents; (d) activity diagram of Cr6+ reduction by 30 mg 0.5%Ti-SnO2/Sn3O4 at different pH values of 10 mg·L-1; (e) 0.5%Ti- SnO2/Sn3O4 cycle experiment

图 11 催化剂样品降解染料活性图: (a) 降解10 mg·L-1 MO; (b) 降解10 mg·L-1 AO-Ⅱ
Figure 11 Degradation activity diagrams of the catalyst samples: (a) degradation of 10 mg·L-1 MO; (b) degradation of 10 mg·L-1 AO-Ⅱ

图 12 Ti-SnO2/Sn3O4光催化去除Cr(Ⅵ)和染料的机理图
Figure 12 Mechanism diagram of Ti-SnO2/Sn3O4 photocatalytic removal of Cr(Ⅵ) and dyes
表 1 样品的比表面积、孔容和孔径
Table 1. Specific surface area, pore volume and pore size of the samples
| Sample | Specific surface area / (m2·g-1) | Pore volume / (cm3·g-1) | Pore size / nm |
| Sn3O4 | 3 | 0.027 8 | 11.25 |
| 0.3%Ti-SnO2/Sn3O4 | 22 | 0.116 9 | 20.54 |
| 0.5%Ti-SnO2/Sn3O4 | 25 | 0.112 0 | 17.32 |
| 0.7%Ti-SnO2/Sn3O4 | 20 | 0.099 7 | 19.07 |
| 0.9%Ti-SnO2/Sn3O4 | 24 | 0.137 5 | 23.59 |
 下载: 导出CSV
下载: 导出CSV
表 2 催化剂样品的间接带隙
Table 2. Indirect band gap of catalyst samples
| Sample | Eg / eV |
| Sn3O4 | 2.77 |
| 0.3%Ti-SnO2/Sn3O4 | 2.76 |
| 0.5%Ti-SnO2/Sn3O4 | 2.59 |
| 0.7%Ti-SnO2/Sn3O4 | 2.52 |
| 0.9%Ti-SnO2/Sn3O4 | 2.42 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们