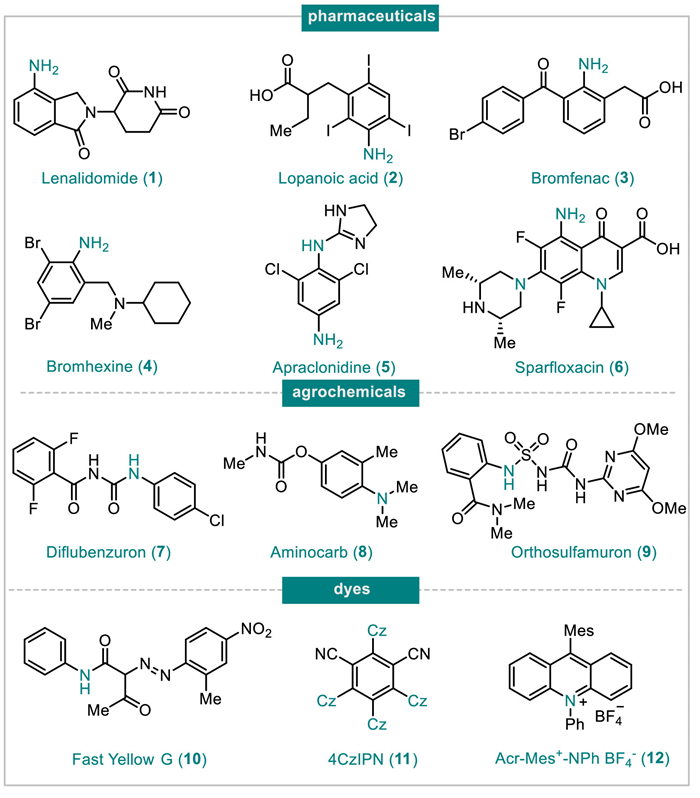
Figure 1.
The representative functional molecules containing the aniline motif (Cz = carbazole, Mes = mesitylene).
Single-atom cobalt catalysts for chemoselective hydrogenation of nitroarenes to anilines
Xiaochun Liu , Chaoyue Wang , Jiaolong Meng , Xiaodong Yue , Qingyun Wang , Jiatian Lu , Junke Wang , Xicun Wang , Yingxiao Zong , Xuefeng Jiang

The anilines are among the most fundamental building block in organic synthesis, and widely assembled in pharmaceuticals, agrochemicals, dyes, etc. (Fig. 1) [1–6]. The current industrial processes of anilines are mainly catalytic hydrogenation of nitroarenes under the H2 atmosphere with transition metals, in which the noble metals of platinum group (such as Pd, Pt, Ru, and Rh) act as mainstream catalysts and have been comprehensively developed in academic laboratories and industries [7–11]. In general, indiscriminate hydrogenation and/or dehalogenation will be conducted under enough catalysts and excess H2 atmosphere when the substrates possess various unsaturated bonds and/or carbon-halogen bonds. In order to improve the chemical selectivity, the precious metal is passivated by attaching it to other metals or metal oxides, but the reaction conditions are harsher, such as higher temperature, longer time, and higher pressure. Indeed, most of the reported noble metal catalysts do not meet the requirements of both catalytic activity and chemical selectivity in the hydrogenation process at the same time [12]. Moreover, the cost of precious metals limits their large-scale applications in the chemical industry.
Recently, the nano crystallization of metal materials has brought about a revolutionary breakthrough in catalysis [13–15]. Further research in the atomic scale of catalytic active centers (single-atomic catalysts, SACs) is springing up to occupy the catalytic highland. SACs possess the maximum atom-utilization efficiency, unique electronic structures, and unconventional catalytic activities, which have attracted wide attention and prompted the development of general synthesis for different applications [16,17]. Moreover, the interaction between isolated metal atoms and solid carriers effectively prevents the atoms from aggregation, and the active sites, geometrical configuration, and highly uniform active centers are analogous to homogeneous catalysts. Meanwhile, SACs are also featured with easy separation, and recycling of heterogeneous catalysts [18–21].
SACs have the advantages of both homogeneous and heterogeneous catalysts. Thus, the development of SACs is considered to narrow the huge gap between homogeneous and heterogeneous catalysts. Indeed, SACs have already been employed in the fields of conventional organocatalysis, photocatalysis, and electrocatalysis [22–26]. Hence, some novel SACs have emerged, in particular, the supported cobalt catalysts showed excellent catalytic activity and chemo-selectivity for the reduction of various functionalized nitroarenes [27–30]. Considering atomic utilization, economic benefit and catalytic efficiency, it is very desirable to improve the catalytic activity and selectivity of cobalt SACs. The groups of Beller [31,32], Gascon [33], and Yang [34] have reported the elegant nitrogen-doped carbon-supported cobalt catalysts (Scheme 1). However, the rapid development of SACs also faces some disadvantages such as high catalyst cost, poor stability, easy aggregation of single atoms, and serious loss of active sites. Therefore, the design of stable and cost-effective single-atom cobalt catalysts remains challenging.
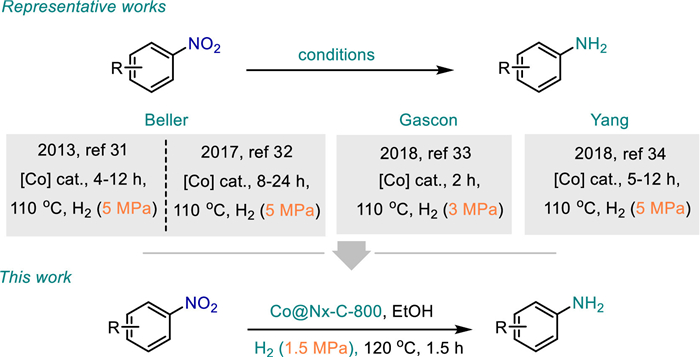
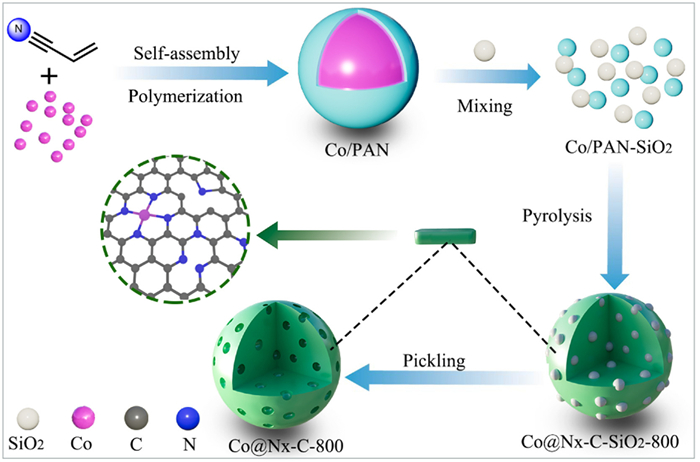
In recent years, we have devoted ourselves to the design, preparation and application of heterogeneous catalysts [35]. Herein, mesoporous nitrogen doped carbon-supported single-atomic cobalt catalysts (Co@Nx-C-800) were prepared by self-assembly polymerization, pyrolysis and pickling using cheap acrylonitrile as monomer and cobalt ion as template. Under mild reaction conditions (393 K, 1.5 MPa H2), Co@Nx-C-800 catalyst showed excellent catalytic performance for the reduction of nitroaromatics to the corresponding aromatic amines, with the conversion and selectivity up to 99%. Moreover, no significant decrease in activity was observed after seven cycles of the catalyst. Experiments and density functional theory (DFT) calculations show that Co-N4 is the most likely catalytic active center in terms of hydrogen splitting, adsorption energy and electronic structure.
Co@Nx-C-800 catalyst was prepared by an acrylonitrile-derived nitrogen-doped carbon loading strategy (Fig. 2). Acrylonitrile and cobalt chloride hexahydrate were used as starting materials. In the presence of AIBN, the blue solid was obtained by reflux in ethyl acetate, which was recorded as Co/PAN. Co/PAN and colloidal-SiO2 were mixed evenly in the solution and the solvent was removed under decompression condition to afford the dark green solid (Co/PAN-SiO2). The carbonization process of Co/PAN-SiO2 was completed by annealing at different temperatures in N2 atmosphere, to provide Co@Nx-C-SiO2-T (T = 700, 800, 900 ℃), followed by the treatment with hydrofluoric acid to obtain Co@Nx-C-T. Nanometer CoNPs@N—C-800 catalyst was prepared by increasing the amount of cobalt chloride hexahydrate and removing silica with 1 mol/L NaOH.
The preliminary structure of the Co@Nx-C-T catalyst was investigated by Fourier-transform infrared (FTIR) spectra (Fig. S1 in Supporting information). For the convenience of analysis, PAN and Co/PAN are used for comparison. The spectra of PAN and Co/PAN are consistent, indicating that the cobalt ions were indeed coated with polyacrylonitrile and that no explicit bond was formed. The FTIR spectra displayed C—H (2940 cm−1), C≡N (2245 cm−1), C=C (1629 cm−1), and C-C (1458 cm−1) in Co/PAN. After the heat treatment, C-H and C≡N disappeared, while C=C (1572 cm−1) shifted to lower wavenumbers due to the conjugative effect in Co@Nx-C-SiO2–800 and Co@Nx-C-800 [36–39]. Co@Nx-C-SiO2–800 contains Si-O-Si (1037 cm−1), which disappeared after pickling, indicating that the 10 wt% HF aqueous solution can wash away the silica. The above results prove that the bond is reconstructed at high carbonization temperature.
The structural change of Co/PAN-SiO2 was monitored by thermogravimetric analysis (TGA) with increasing pyrolysis temperature in the nitrogen atmosphere (Fig. S2 in Supporting information). Weight loss prior to 230 ℃ was due to the physically absorbed water. Subsequently, the mass loss at 300–500 ℃ was ascribed to the conversion of C≡N to C=N and C=C groups, along with releasing HCN, NH3, and CH4, resulting in extreme mass loss during this stage. When the temperature reaches 650 ℃, the mass was further decreased, which indicated that the carbon skeleton may be broken and decomposed [40,41]. Moreover, the carbon structures of Co@Nx-C-T catalysts were studied by Raman spectra (Fig. S3 in Supporting information). D and G bands of the catalysts are located at 1350 cm−1 and 1580 cm−1, respectively. D bands are commonly attributed to disordered carbon structures. G bands can be attributed to sp2 hybrid carbon atoms. The intensity ratios of ID/IG reduce with the increase of pyrolysis temperature, which indicates that the degree of graphitization of the catalysts increased with the increase of pyrolysis temperature, resulting in the reduction of defect sites.
X-ray diffraction (XRD) patterns of the Co@Nx-C-SiO2–800, N—C-800, Co@Nx-C-T, and CoNPs@N—C-800 are also presented (Fig. 3a). Co@Nx-C-SiO2–800 sample shows a broad diffraction peak at 2θ = 22.4°, which is assigned to amorphous silica. However, the broad diffraction peak is not observed in Co@Nx-C-T (T = 700, 800, 900 ℃), indicating that the 10 wt% HF aqueous solution can etch silica. N—C-800 in XRD patterns shows two prominent peaks at 26.2° and 44.0°, corresponding to the (002) and (101) planes of graphitic carbon [42]. Meanwhile, the diffraction peaks of Co@Nx-C-800 are similar to N—C-800. Therefore, we conclude that no characteristic peaks of Co-based crystals are found in Co@Nx-C-800, which shows cobalt species were highly dispersed in carbon carriers. However, there are three diffraction peaks in CoNPs@N—C-800, which appeared at θ = 44.2°, 51.5° and 75.9°, respectively, corresponding to the (111), (200) and (220) planes of cobalt nanoparticles (Fig. 3a) [43]. The results indicate that cobalt species in CoNPs@N—C-800 catalysts mainly exists in the form of cobalt nanoparticles.
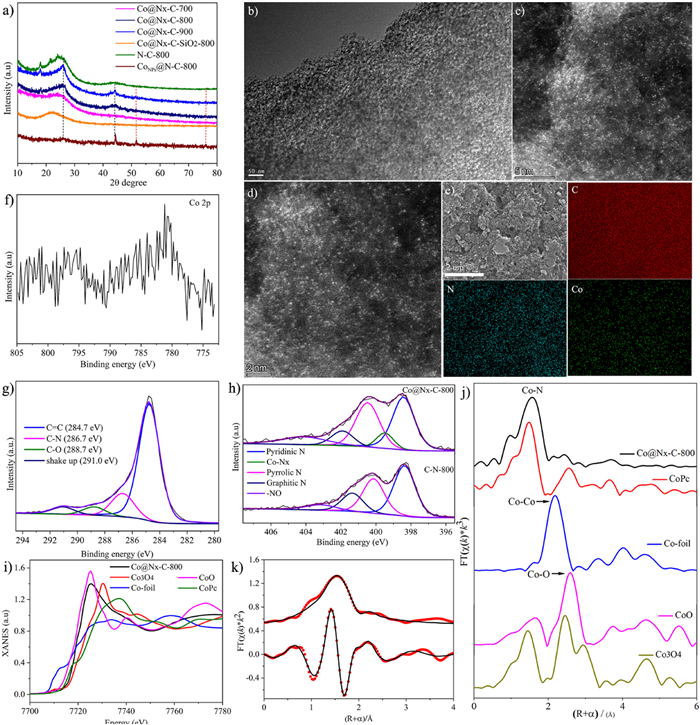
As shown in Fig. 3b and Fig. S4 (Supporting information), the structure of Co@Nx-C-T was characterized by transmission electron microscopy (TEM). The porous structures were clearly presented. No cobalt clusters or nanoparticles in Co@Nx-C-800 was observed by aberration-corrected HAADF-STEM, which demonstrated Co species are atomically dispersed in carbon carriers (Figs. 3c and d). Moreover, N and Co atoms are homogeneously distributed in the carbon matrices (Fig. 3e). The cobalt content of Co@Nx-C-800 was determined by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) to be 1.10%. In order to gain a deeper insight into the pore structures, Nitrogen adsorption-desorption isotherms were performed to investigate the porosities of Co@Nx-C-T (Figs. S5 and S6 in Supporting information). Nitrogen adsorption isotherms of Co@Nx-C-T showed a type Ⅳ curve and an H2-type hysteresis loop, characteristic of a mesoporous structure. According to the Brunauer-Emmett-Teller method, the surface area of Co@Nx-C-T (T = 700, 800, 900 ℃) was determined to be 317.25, 349.43, and 487.55 m2/g, respectively. The Barrett-Joyner-Halenda average pore size and pore volume were determined to be 13.93, 13.78, 13.43 nm, and 0.855, 1.124, and 1.280 cm3/g, respectively. With the increase of temperature, the specific surface area and pore volume increase, which is conducive to the exposure of active sites and accelerate the reduction reaction (see Table S1 for the detailed data).
The valence states of cobalt, carbon, and nitrogen in Co@Nx-C-800 have been investigated by X-ray photoelectron spectroscopy (XPS). Due to the very low cobalt content in Co@Nx-C-800, the peak of Co 2p is so weak that it is impossible to accurately judge the valence state of Co (Fig. 3f). C 1s XPS can be deconvoluted into four peaks: C=C (284.7 eV), C-O (286.7 eV), C-N (288.7 eV), and shake-up carbon (291.0 eV) (Fig. 3g and Fig. S7 in Supporting information) [44,45]. N 1s XPS can be deconvoluted into five peaks: pyridine N (398.4 eV), Co-Nx (399.5 eV), pyrrole N (400.5 eV), graphite N (401.7 eV) and -NO (404.1 eV). However, N 1s XPS spectrum of C-N-800 is only deconvoluted into four peaks (pyridine N, pyrrole N, graphite N and -NO), suggesting that Co-Nx is the active site in Co@Nx-C-800 (Fig. 3h) [46–48]. Meanwhile, there is no Co-Nx peak in Co@Nx-C-700, leading to the low catalytic activity (Fig. S8 in Supporting information).
The electronic and coordination structures of cobalt atoms were examined by X-ray absorption near side structures (XANES) and extended X-ray absorption fine structures (EXAFS). XANES shows the energy absorption threshold of Co@Nx-C-800 situated between CoO and Co3O4, elucidating that the Co atoms in the catalyst carry positive charges and the valence state of Co atoms is between +2 and +3 (Fig. 3i). Fourier-transformed (FT) k3-weighted EXAFS spectra of Co@Nx-C-800 show one main peak at about 1.5 Å, corresponding to the Co-N first coordination. Meanwhile, comparing with Co foil, CoO, Co3O4, and CoPc (Cobalt phthalocyanine), the peak of Co-Co (2.19 Å) metal bond is not observed, notarizing the successful synthesis of atomically dispersed Co single-atomic catalysts (Fig. 3j). It is in consistent with the results of the HAADF-STEM. The EXAFS fitting results indicate that the Co-N coordination number is 4.2 with a bond length of 2.06 Å (Fig. 3k and Fig. S9 in Supporting information). This indicates that the Co atom was coordinated by four nitrogen atoms (see Table S2 in Supporting information for the detailed data).
The DFT calculation was performed to further verify the value of x in the catalytic active site Co-Nx. We studied the dissociation of H2 on perfect Co@graphene (Co@GP) and Co@N-doped graphene (Co@xNGP, x = 1, 2, 3, 4) respectively (Fig. S10 in Supporting information). Both Co@1NGP and Co@2NGP catalysts have little effect on the dissociation of H2, giving a close activation energy of about 1.1 eV. Once the NGP is doped on the Co atom to form Co@3NGP and Co@4NGP, the activation energy is significantly reduced, which implies that Co-N can improve the activity in H2 activation with the activation energies of 0.93 and 0.89 eV. To elaborate the formation of the Co-N bond can enhance the catalytic activity. The adsorption energy of Co@GP and Co@xNGP are explored in Fig. S11 (Supporting information). The adsorption energies of Co@GP, Co@1NGP, and Co@2NGP are relatively close, while the adsorption energies of Co@3NGP and Co@4NGP increase significantly, indicating that the stability of Co@4NGP is optimal. The above facts indicate that Co-N4 is the most likely catalytic active center. To illustrate the nature of the improved catalytic activity, the electronic structures of the catalysts are investigated. As for Co@GP, Co@1NGP, and Co@2NGP, there is an electron transfer of net 0.13–0.17 e from the adsorbed Co atom to GP and NGP (Fig. S12 in Supporting information). When Co is doped into NGP to form Co-3NGP and Co@4NGP, more electrons are transferred from Co to N due to the more electronegative of N, making the Co more positive charge-rich, which would be beneficial to active H2 because the 3d empty orbit of Co is easier to receive electrons.
To inspect the relationship between the structure and the performance of the catalyst, 4-chloronitrobenzene was selected as the template material to investigate the catalytic performance of different cobalt materials. Both Co@Nx-C-800 and Co@Nx-C-900 showed excellent catalytic performance with 100% conversion and 99% selectivity, while Co@Nx-C-700, CoCl2‧6H2O, Co/PAN, and N—C-800 as catalysts had poor catalytic effect, or even could not catalyze the reaction (Table 1, entries 1–6). These facts indicated that Co-Nx may be the active site of hydrogenation reaction, and simple cobalt ions or weakly coordinated cobalt ions have no effective catalytic activity. The lower catalytic activity of Co@Nx-C-700 may be due to the absence of Co-Nx. This conclusion is supported by XPS. Meanwhile, the Co@Nx-C-SiO2–800 catalyst also has very weak activity, indicating that the pore effect is vital on the activity (entry 7). In order to further demonstrate the high reactivity of the single-atom Co@Nx-C-800 catalyst, nano CoNPs@N—C-800 catalyst (Co content 17.2%) was used to catalyze the reaction with conversion was only 50.4% (entry 8). In order to verify the importance of hydrogen, we performed controlled experiments and found that the reaction did not occur in nitrogen atmosphere (entry 9). In general, the dehalogenated byproducts are usually produced in the hydrogenation of halogenated nitroarenes by traditional noble metal catalysts [49]. Notably, no such dehalogenated byproducts were observed in gas chromatography and NMR using the developed monoatomic cobalt catalysts. Compared with the previously reported catalysts [31–34], Co@Nx-C-800 and Co@Nx-C-900 showed excellent activity under milder reaction conditions.
Since the yield of Co@Nx-C-900 is relatively low due to higher temperature in the synthesis process, Co@Nx-C-800 was selected as the catalyst for further optimization. In most of tested solvents, high conversion and selectivity were achieved under 1.5 MPa hydrogen gas at 120 ℃ for 1.5 h (Table 2, entries 1–3, 5, 6). However, the conversion rate is greatly reduced in THF, which may be due to the oxygen atoms in THF binding to the active center, resulting in reduced catalytic activity (entry 4). In addition, it was found in the experiment that THF can make the catalyst caked, which may also lead to the deactivation of the catalyst. Notably, environmentally benign and green solvent, water, can successfully lead to efficient catalysis with good selectivity. Decreasing hydrogen pressure and temperature led to lower yields with retaining the high selectivity in EtOH (entries 7–11).
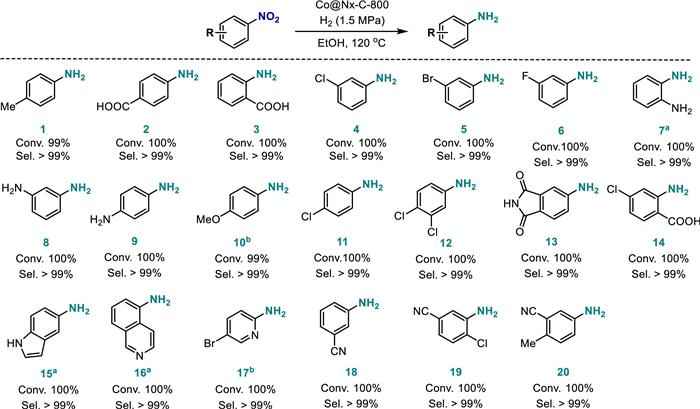
With the optimized reaction conditions in hand, we investigated the generality of Co@Nx-C-800 in the catalytic hydrogenation of aromatic nitro substrates with different substituents. The results show that the catalyst had high activity and selectivity for nitroaromatics with electron-donating or electron-withdrawing groups (Scheme 2). In addition to the high activity of the Co@Nx-C-800, the chemoselective reduction of nitro compounds is also very important. As shown in Scheme 2, in the presence of halogen and nitrile group, Co@Nx-C-800 had high chemical selectivity for the hydrogenation of substituted nitroaromatics (4–6, 11, 12, 14, 17–20). Furthermore, the catalyst also had good performance for heterocyclic nitroarenes in a longer reaction time (15–17). The reusability of the catalyst was examined upon the reduction of 4-chloronitrobenzene with H2 (Fig. S13 in Supporting information). It was found that the conversion rate of 4-chloronitrobenzene was still maintained at > 98%, and the chemo-selectivity was maintained at > 99% after seven times recycling. The cobalt content of spent SACs Co@Nx-C-800 was 0.79% by ICP-OES detection. The catalyst loses a total of 0.31% of cobalt element after seven cycles times, indicating that the Co@Nx-C-800 has less active site loss in the recycling experiment and a good stability and durable structure in the liquid reaction. All of the facts show that the catalyst is very promising and encouraging from a practical point of view.
In summary, a novel SAC (Co@Nx-C-800) with excellent catalytic activity and selectivity has been developed for the reduction of nitroarenes to anilines. Moreover, it shows outstanding stability and reusability without obvious activity loss after recycling seven times, which provides a versatile strategy to introduce various metal species on nitrogen-doped carbon with targeted and improved properties for diverse catalytic reactions. Further SACs catalysts are under investigation in our lab to achieve large-scale processes in the chemical industry.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 22061017 and 21862006), Science and technology program of Gansu Province (Nos. 22YF7GG127 and 23JRRG0002), Hexi University Research Start-up Fund Project (Nos. KYQD2020013). We also thank Prof. Qingyun Wang for the density functional theory of the materials.
Supplementary material associated with this article can be found, in the online version, at doi:
F.C. Lizana, S.G. Quero, M.A. Keane, ChemSusChem 1 (2008) 215–221. doi: 10.1002/cssc.200700105
A.M. Tafesh, J. Weiguny, Chem. Rev. 96 (1996) 2035–2052. doi: 10.1021/cr950083f
S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, 2001.
J.P. Adams, J.R. Paterson, J. Chem. Soc., Perkin Trans. 1 (2000) 3695–3705.
Z. Li, C. Han, Chin. Chem. Lett. 31 (2020) 818–820. doi: 10.1016/j.cclet.2019.06.047
X. Xie, Z. Wu, N. Zhang, Chin. Chem. Lett. 31 (2020) 1014–1017. doi: 10.1016/j.cclet.2019.10.012
J. Zhou, Y. Li, H.B. Sun, et al., Green Chem. 19 (2017) 3400–3407. doi: 10.1039/C7GC00986K
D. Xu, F. Wang, G. Yu, et al., ChemCatChem 10 (2018) 4569–4577. doi: 10.1002/cctc.201800987
A.N. Chishti, Z. Ma, J. Zha, et al., Chin. Chem. Lett. 34 (2023) 108122. doi: 10.1016/j.cclet.2022.108122
Q. Wei, Y.S. Shi, K.Q. Sun, B.Q. Xu, Chem. Commun. 52 (2016) 3026–3029. doi: 10.1039/C5CC07474F
Z. Fang, Y. Gu, X. Dong, et al., Chin. Chem. Lett. 34 (2023) 108128. doi: 10.1016/j.cclet.2022.108128
U. Siegrist, P. Baumeister, H. Blaser, M. Studer, Chemical Industries, Marcel Dekker, New York, 1998.
S. Mitchell, R. Qin, N. Zheng, P.R. Javier, Nat. Nanotechnol. 16 (2021) 129–139. doi: 10.1038/s41565-020-00799-8
M. Sankar, Q. He, R.V. Engel, et al., Chem. Rev. 120 (2020) 3890–3938. doi: 10.1021/acs.chemrev.9b00662
V.C. Marta, M. Marta, F.S. Jesus, et al., Acc. Chem. Res. 53 (2020) 520–531. doi: 10.1021/acs.accounts.9b00609
R. Lang, X. Du, Y. Huang, et al., Chem. Rev. 120 (2020) 11986–12043. doi: 10.1021/acs.chemrev.0c00797
H. Zhou, Y. Zhao, J. Xu, et al., Nat. Commun. 11 (2020) 335. doi: 10.1038/s41467-019-14223-w
R. Lang, T. Li, D. Matsumura, et al., Angew. Chem. Int. Ed. 55 (2016) 16054–16058. doi: 10.1002/anie.201607885
X. Cui, W. Li, P. Ryabchuk, K. Junge, M. Beller, Nat. Catal. 1 (2018) 385–397. doi: 10.1038/s41929-018-0090-9
S. Ji, Y. Chen, X. Wang, et al., Chem. Rev. 120 (2020) 11900–11955. doi: 10.1021/acs.chemrev.9b00818
L. Wang, W. Zhang, S. Wang, et al., Nat. Commun. 7 (2016) 14036. doi: 10.1038/ncomms14036
B. Chen, X. Zhong, G. Zhou, N. Zhao, H.M. Cheng, Adv. Mater. 34 (2022) 2105812. doi: 10.1002/adma.202105812
W. Guo, Z. Wang, X. Wang, Y. Wu, Adv. Mater. 33 (2021) 2004287. doi: 10.1002/adma.202004287
X. Deng, B. Qin, R. Liu, et al., J. Am. Chem. Soc. 143 (2021) 20898–20906. doi: 10.1021/jacs.1c09535
F. Estefanía, A. Miguel, C. Rivero, et al., J. Am. Chem. Soc. 141 (2019) 1928–1940. doi: 10.1021/jacs.8b07884
Z. Li, W. Wei, H. Li, et al., ACS Nano 15 (2021) 10175–10184. doi: 10.1021/acsnano.1c02094
G. Zhang, F. Tang, X. Wang, L. Wang, Y.N. Liu, ACS Catal. 12 (2022) 5786–5794. doi: 10.1021/acscatal.2c01113
H. Jin, P. Li, P. Cui, et al., Nat. Commun. 13 (2022) 723. doi: 10.1038/s41467-022-28367-9
L. Zhang, N. Shang, S. Gao, et al., ACS Catal. 10 (2020) 8672–8682. doi: 10.1021/acscatal.0c00239
M. Li, C. Zhang, Y. Tang, et al., ACS Catal. 12 (2022) 11960–11973. doi: 10.1021/acscatal.2c03200
F.A. Westerhaus, R.V. Jagadeesh, G. Wienhöfer, et al., Nat. Chem. 5 (2013) 537–543. doi: 10.1038/nchem.1645
D. Formenti, F. Ferretti, C. Topf, et al., J. Catal. 351 (2017) 79–89. doi: 10.1016/j.jcat.2017.04.014
X. Sun, A.I.O. Suarez, D. Osadchii, et al., J. Catal. 357 (2018) 20–28. doi: 10.1016/j.jcat.2017.10.030
T. Song, P. Ren, Y. Duan, et al., Green Chem. 20 (2018) 4629–4637. doi: 10.1039/C8GC01374H
J. Wang, Y. Zong, X. Wang, et al., Green Chem. 18 (2016) 967–973. doi: 10.1039/C5GC01551K
N. Grassie, R. Mcguchan, Eur. Polym. J. 8 (1972) 257–269. doi: 10.1016/0014-3057(72)90032-8
I. Shimada, T. Takahagi, M. Fukuhara, K. Morita, A. Ishitani, J. Polym. Sci., Part A: Polym. Chem. 24 (1986) 1989–1995. doi: 10.1002/pola.1986.080240819
C.R. Wu, B. Iedberg, J. Polym. Sci., Part B: Polym. Phys. 26 (1988) 1127–1136. doi: 10.1002/polb.1988.090260516
J.L. Brédas, J. Chem. Phys. 85 (1986) 2219–2226. doi: 10.1063/1.451116
R. Zheng, Z. Mo, S. Liao, et al., Carbon 69 (2014) 132–141. doi: 10.1016/j.carbon.2013.11.075
W.M. Millan, T.T. Thompson, L.G. Arriaga, M.A. Smit, Int. J. Hydrogen Energy 34 (2009) 694–702. doi: 10.1016/j.ijhydene.2008.11.006
Z.Y. Wu, M. Karamad, X. Yong, et al., Nat. Commun. 12 (2021) 2870. doi: 10.1038/s41467-021-23115-x
P. Zhou, L. Jiang, F. Wang, et al., Sci. Adv. 3 (2017) e1601945. doi: 10.1126/sciadv.1601945
J.M. Stillahn, K.J. Trevino, E.R. Fisher, ACS Appl. Mater. Interfaces 3 (2011) 1402–1410. doi: 10.1021/am101282y
H.H. Wang, N. Leaukosol, Z.B. He, et al., Cellulose 20 (2013) 1587–1601. doi: 10.1007/s10570-013-9945-z
X. Li, Y.H. He, S. Cheng, et al., Adv. Mater. 33 (2021) 2106371. doi: 10.1002/adma.202106371
D.W. Xi, J.Y. Li, J.X. Low, et al., Adv. Mater. 34 (2022) 2104090. doi: 10.1002/adma.202104090
M.H. Li, H.F. Wang, W. Luo, et al., Adv. Mater. 32 (2020) 2001848. doi: 10.1002/adma.202001848
Y. Pan, R. Lin, Y. Chen, et al., J. Am. Chem. Soc. 140 (2018) 4218–4221. doi: 10.1021/jacs.8b00814

Figure 1 The representative functional molecules containing the aniline motif (Cz = carbazole, Mes = mesitylene).

Figure 3 (a) XRD patterns of Co@Nx-C-700, Co@Nx-C-800, Co@Nx-C-900, Co@Nx-C-SiO2–800, C—N-800 and CoNPs@N—C-800. (b) TEM images of Co@Nx-C-800. (c, d) Aberration-corrected HAADF-STEM image of Co@Nx-C-800. (e) Elemental mapping image of Co@Nx-C-800. (f) Co 2p XPS spectrum of Co@Nx-C-800. (g) C 1s XPS spectrum of Co@Nx-C-800. (h) N 1s XPS spectra of Co@Nx-C-800 and C—N-800. (i) XANES of Co K-edge. (j) FT at the Co K-edge of Co@Nx-C-800, CoO, CoPc, Co-foil, and Co3O4. (k) Co K-edges EXAFS (points) and the curve-fit (line) for Co@Nx-C-800, shown in R-space (FT-magnitude and imaginary component).

Scheme 2 Substrate scope. Reaction conditions: nitroarenes (1 mmol), Co@Nx-C-800 (40 mg), ethanol (10 mL), 120 ℃; H2 1.5 MPa, 1.5 h. a 10 h, b 4 h. The con. and sel. were determined using GC.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:

