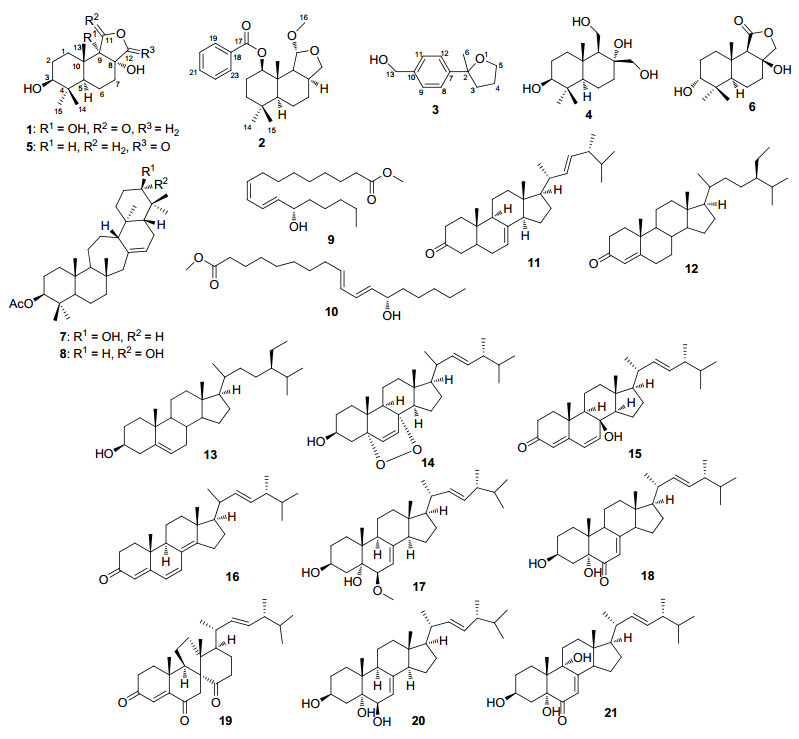
 图 1
化合物1~21结构图
Figure 1.
Structures of compounds 1~21
图 1
化合物1~21结构图
Figure 1.
Structures of compounds 1~21
引用本文:
杨宁宁, 孔凡栋, 马青云, 黄圣卓, 罗都强, 周丽曼, 戴好富, 郁志芳, 赵友兴. 多形炭角菌Xylaria polymorpha菌丝发酵的次级代谢产物研究[J]. 有机化学,
2017, 37(4): 1033-1039.
doi:
10.6023/cjoc201612039

Citation: Yang Ningning, Kong Fandong, Ma Qingyun, Huang Shengzhuo, Luo Duqiang, Zhou Liman, Dai Haofu, Yu Zhifang, Zhao Youxing. Chemical Constituents from the Cultures of Fungus Xylaria polymorpha[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 1033-1039. doi: 10.6023/cjoc201612039
Citation: Yang Ningning, Kong Fandong, Ma Qingyun, Huang Shengzhuo, Luo Duqiang, Zhou Liman, Dai Haofu, Yu Zhifang, Zhao Youxing. Chemical Constituents from the Cultures of Fungus Xylaria polymorpha[J]. Chinese Journal of Organic Chemistry, 2017, 37(4): 1033-1039. doi: 10.6023/cjoc201612039
多形炭角菌Xylaria polymorpha菌丝发酵的次级代谢产物研究
摘要:
从多形炭角菌Xylaria polymorpha (Pers.:Fr.)Grer菌丝发酵产物乙酸乙酯部分中分离鉴定了21个化合物,包括2个新的drimane型倍半萜polymorphines A,B(1~2),1个新的phenyloxolane类化合物2-甲基-2-(4-羟甲基苯基)氧杂环戊烷(3)和18个已知化合物4~21.利用NMR和X射线单晶衍射技术对上述化合物结构进行鉴定与确认.化合物2具有一定的乙酰胆碱酯酶及α-葡萄糖苷酶抑制活性.此外,化合物3具有中等的全齿复活线虫Panagrellus redivivus抑制活性,在浓度为2.5 mg/mL时,线虫致死率为59.6%.
English
Chemical Constituents from the Cultures of Fungus Xylaria polymorpha
Abstract:
Totally 21 compounds were isolated from the EtOAc extract of fermentation broth of fungus Xylaria polymorpha (Pers.: Fr.) Grer, including two new drimane-type sesquiterpenoids named polymorphines A and B (1~2) and one new phenyloxolane compound named 2-methyl-2-(4-hydroxymethylphenyl) oxolane (3), together with 18 known compounds 4~21. The structures of these compounds were elucidated by NMR and single-crystal X-ray diffraction analysis. Compound 2 exhibited anti-acetylcholinesterase and α-glucosidase inhibitory activities. Moreover, compound 3 showed moderate inhibitory activitiy against the nematode Panagrellus redivivus with mortality ratio of 59.6% at 2.5 mg/mL.
-
多形炭角菌Xylaria polymorpha (Pers.: Fr.) Grer为炭角菌属的高等真菌, 子实体较小, 呈炭棒状, 质地较为坚硬, 主要分布于中国的海南、广东、福建、台湾等东南地区.该真菌主要生长于林间的腐木、树皮的裂缝中.现有的研究报道表明多形炭角菌具有丰富的化合物类型及活性, 比如具有ABTS自由基清除活性的恶庚因类化合物[1], 具有抗肿瘤活性的二萜糖苷[2], 以及具有抗真菌活性的聚丙酸酯类等[3].
由于多形炭角菌的资源比较匮乏, 难以大量收集, 本课题组研究其菌丝的发酵产物, 以期发现具有显著药理活性的化合物.通过多种色谱分离技术对其菌丝发酵产物的乙酸乙酯部分进行纯化, 获得21个单体化合物, 并利用NMR、HRESIMS、X射线单晶衍射、紫外和红外等波谱技术对其结构进行分析, 分别鉴定为2个新的倍半萜polymorphines A, B (1, 2), 1个新的phenyloxolane化合物2-甲基-2-(4-羟甲基苯基) 氧杂环戊烷 (3), 和18个已知化合物agripilol A (4)[4], 3β-hydroxy-peniopholide (5)[5], sulphureuine E (6)[6], 21-epi-serratenediol-3-acetate (7)[7], serratenediol-3-acetate (8)[8], (-)-(13S, 9Z, 11E)-13-羟基-9, 11-十八碳二烯酸甲酯 (9)[9], (±)-methyl-13-hydroxy-9E, 11E-octadecadienoate (10)[10], 麦角甾-7, 22-二烯-3-酮 (11)[11], β-谷甾酮 (12)[12], β-谷甾醇 (13)[13], (22E)-5α, 8α-表二氧麦角甾-6, 22-二烯-3β-醇 (14)[14], isocyathisterol (15)[15], 麦角甾-4, 6, 8 (14), 22-四烯-3-酮 (16)[16], (3β, 5α, 6β, 22E)-6-甲氧基麦角甾-7, 22-二烯-3, 5-二醇 (17)[17], 3β, 5α-二羟基-(22E, 24R)-麦角甾-7, 22-二烯-6-酮 (18)[18], dankasterone A (19)[19], (22E, 24R)-麦角甾-7, 22-二烯-3β, 5α, 6β-三醇 (20)[20]和 (22E, 24R)-麦角甾-7, 22-二烯-3β, 5α, 9α-三醇-6-酮 (21)[20] (图 1).我们对上述化合物进行了AChE抑制活性, α-葡萄糖苷酶活性及抗线虫活性的筛选.结果显示化合物2具有一定的乙酰胆碱酯酶及α-葡萄糖苷酶抑制活性.此外, 化合物3具有中等的全齿复活线虫Panagrellus redivivus抑制活性, 在浓度为2.5 mg/mL时, 线虫致死率为59.6%.本文对上述化合物的分离纯化、结构鉴定及生物活性进行详细报道.
图 1
化合物1~21结构图
Figure 1.
Structures of compounds 1~21
1 结果与讨论
1.1 化合物的结构鉴定
化合物1:白色无定型粉末, 易溶于甲醇, 高分辨质谱 (HRESIMS) 显示化合物的准分子离子峰为307.1517 [M+Na]+(calcd for C15H24O5Na, 307.1516), 推断其分子式为C15H24O5, 不饱和度为4. IR显示化合物中有羟基 (3384 cm-1) 和内酯羰基 (1765 cm-1) 的存在.分析其1H NMR (表 1) 数据, 推断出1含有3个单峰甲基信号[δH 0.85 (s, 3H, H-13), 1.26 (s, 3H, H-14), 1.04 (s, 3H, H-15)]. 13C NMR谱和DEPT谱给出了15个碳信号, 包括3个甲基、5个亚甲基 (1个含氧)、2个次甲基和5个季碳 (1个羰基和2个含氧).以上核磁数据显示化合物1在结构上与该种分离鉴定的化合物sulphureuine E (6) 十分相似, 都为具有6/6/5三环体系的倍半萜, 区别在于化合物1在C-9 (dC 79.6) 位上多了1个羟基.在HMBC谱图中, H2-12 [dH 4.52 d (8.5), 3.95 d (8.5)]和H3-13 [dH (s, 0.85)]都与季碳C-9相关, 进一步证实C-9被羟基化.化合物1的相对构型可以通过分析ROESY谱图确定.在ROESY谱图 (图 3) 中, H-5 [dH 1.61 dd (3.0, 13.0)]和H-3 [dH 3.24 dd (6.2, 10.0)], H3-15 [dH 1.04, s]及H-7α [dH 1.98, m]相关, 表明以上氢均位于α位, 而H3-14 [dH 1.26, s]/H3-13[dH 0.85, s]/H-6β[dH 1.66, m]/H-12β[dH 4.52 d (8.5)]/H-7β [dH 2.0, m]等NOE效应说明H3-14、H3-13、H-6β、H-12β和H-7β都是处于β位, 进一步推断出OH-8和OH-9一定处于α位.因此, 化合物1被确定为polymorphine A (图 1).
 表 1
化合物1~3的NMR数据
Table 1.
NMR data of 1~3 (1 in CD3OD, 2 and 3 in CDCl3)
表 1
化合物1~3的NMR数据
Table 1.
NMR data of 1~3 (1 in CD3OD, 2 and 3 in CDCl3)
No. 1 2 3 δC δH (J in Hz) δC δH (J in Hz) δC δH (J in Hz) 1 30.0 (t) 2.09, m 1.48, m 85.1 (d) 4.90, dd (4.3, 11.5) 2 26.5 (t) 1.76, m 1.72, m 24.5 (t) 1.78, m 1.74, m 84.3 (s) 3 78.5 (d) 3.24, dd (6.2, 10.0) 39.7 (t) 1.52, m 1.50, m 39.6 (t) 2.19, m 2.02, m 4 39.3 (s) 33.1 (s) 25.9 (t) 1.99, m 1.80, m 5 45.9 (d) 1.61, dd (3.0, 13.0) 51.4 (d) 1.02, dd (3.4, 11.5) 67.7 (t) 4.01, m 3.90, m 6 17.4 (t) 1.66, m 1.62, m 17.7 (t) 1.50, m 1.42, m 29.8 (q) 1.52, s 7 28.2 (t) 2.00, m 1.98, m 24.1 (t) 1.64, m 1.60, m 147.9 (s) 8 78.8 (s) 34.7 (d) 2.56, m 127.1 (d) 7.32, d (8.0) 9 79.6 (s) 57.4 (d) 1.91, d (8.0) 125.1 (d) 7.37, d (8.0) 10 39.1 (s) 39.1 (s) 139.0 (s) 11 174.4 (s) 108.4 (d) 5.03, s 125.1 (d) 7.37, d (8.0) 12 78.4 (t) 4.52, d (8.5) 3.95, d (8.5) 71.7 (t) 3.91, dd (8.3, 9.0) 3.53, dd (8.3, 9.0) 127.1 (d) 7.32, d (8.0) 13 15.4 (q) 0.85, s 12.4 (q) 1.20, s 65.3 (t) 4.67, s 14 17.6 (q) 1.26, s 33.0 (q) 0.92, s 15 28.5 (q) 1.04, s 22.3 (q) 0.92, s 16 54.1 (q) 2.69, s 17 165.8 (s) 18 130.8 (s) 19/23 129.7 (d) 8.05, d (8.0) 20/22 128.5 (d) 7.44, t (7.9) 21 133.0 (d) 7.55, t (7.4) 表 1 化合物1~3的NMR数据
Table 1. NMR data of 1~3 (1 in CD3OD, 2 and 3 in CDCl3)化合物2:白色晶体, 易溶于氯仿, HRESIMS显示化合物的分子式为C23H32O4 (calcd for C23H32O4Na [M+Na]+ 395.2193, found 395.2190), 不饱和度为8.红外光谱IR显示结构中存在羰基 (1719 cm-1) 和芳环 (1610和1585 cm-1).在1H NMR和13C NMR谱图的低场区 (dC/H129.7/8.05, 128.5/7.44, 133.0/7.55, 130.8) 和 (dC 165.8) 显示出结构中含有1个单取代苯环和1个共轭的酯羰基.在HMBC中, 芳香质子信号H-19/H-23与酯羰基C-17相关也证明了结构中存在1个苯甲酰基. 13C NMR谱图的高场区给出了16个碳信号, 包括4个甲基、5个环内亚甲基、5个次甲基及2个季碳.除了甲氧基信号之外, 剩余的15个碳信号与化合物1非常相似, 表明2也是一个drimane型倍半萜的衍生物.最后, 通过1H-1H COSY和HMBC谱图 (图 2) 的综合分析得到化合物2的平面结构.在ROESY谱图中 (图 3), H-13 [dH 1.20, s]和H-15 [dH 0.92, s], H-11 [dH 5.03, s] ROESY信号相关, 表明H-13和H-15, H-11均处于β位, 而H-1 [4.90, dd (4.3, 11.5)]/ H-9 [1.91, d (8.0)]、H-5 [1.02, dd (3.4, 11.5)]/H-9/H-8 [2.56, m]、以及H-5/H-14 [0.92, s]的信号相关, 说明H-1/H-9/ H-5/H-8/H-14处于α位.此外, 我们通过Mo靶X衍射分析进一步确证了化合物2的相对构型 (图 4).综上所述, 化合物2的结构被命名为polymorphine B.
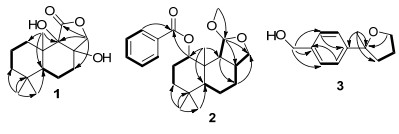 图 2
化合物1~3的主要COSY和HMBC相关信号
Figure 2.
Key 1H-1H COSY and HMBC correlations of compounds 1~3
图 2
化合物1~3的主要COSY和HMBC相关信号
Figure 2.
Key 1H-1H COSY and HMBC correlations of compounds 1~3
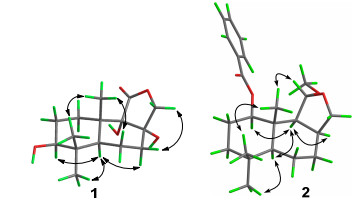 图 3
化合物1和2的主要ROESY相关信号
Figure 3.
Key ROESY correlations of compounds 1and 2
图 3
化合物1和2的主要ROESY相关信号
Figure 3.
Key ROESY correlations of compounds 1and 2
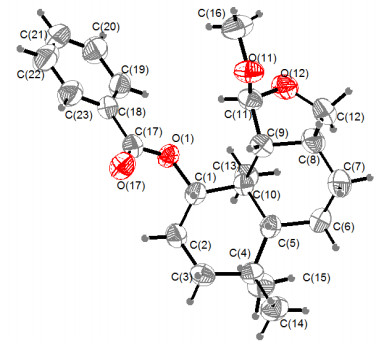 图 4
化合物2的X单晶衍射图
Figure 4.
X-ray crystallography of compound 2
图 4
化合物2的X单晶衍射图
Figure 4.
X-ray crystallography of compound 2
化合物3:淡黄色油状液体, 高分辨质谱 (HRESIMS) 给出了化合物的分子式C12H16O2(测量值215.1045 [M+Na]+, 计算值为215.1043), 不饱和度为5.在红外光谱中, 3444 cm-1处吸收带显示化合物存在羟基.分析1H NMR (表 1) 数据, 推知化合物中有1个单峰甲基信号[δ 1.52 (s, 3H, H-6)]. 13C NMR谱和DEPT谱上显示12个碳信号, 包括1个甲基、4个亚甲基 (2个含氧)、4个芳香族次甲基、3个季碳.比对核磁数据, 发现化合物3和2-甲基-2-(4-甲基苯基) 氧杂环戊烷[21]在结构上非常相似.区别仅在于后者C-13位上的甲基被3中的羟甲基[δC 65.3 (t, C-13), δH 4.67 (s, H-13)]取代.通过进一步分析HMBC和1H-1H COSY谱图, 得到3的平面线性结构 (图 1), 最后命名为2-甲基-2-(4-羟甲基苯基) 氧杂环戊烷.
1.2 化合物的活性测试
对化合物1~6进行了AChE抑制活性和α-葡萄糖苷酶活性的筛选, 结果显示化合物2具有一定的AChE抑制活性 (抑制率为34.3%, 反应终浓度为50 µg/mL), 同时也显示出微弱的α-葡萄糖苷酶抑制活性 (IC50 543.8 µmol/L).对化合物1~8进行了全齿复活线虫Panagrellus redivivus的活性评价, 只有化合物3显示出一定的杀虫活性, 在浓度为2.5 mg/mL时, 对线虫的致死率为59.6%.
2 结论
从多形炭角菌Xylaria polymorpha 的菌丝发酵产物中分离鉴定了21个单体化合物, 包括2个新的drimane型倍半萜、1个新的phenyloxolane类化合物以及18个已知化合物.活性测试结果表明化合物2显示出一定的AChE抑制活性及弱的α-葡萄糖苷酶抑制活性, 化合物3对线虫有致死作用, 具有一定的杀线虫活性.通过本实验及相关文献报道可知多形炭角菌及其菌丝发酵产物的化合物种类和活性比较多样化, 具有进一步研究的价值与意义.
3 实验部分
3.1 仪器与试剂
旋光仪Rudolph Autopol Ⅲ型 (美国鲁道夫公司); 紫外光谱仪 (Beckman Shimadzu UV-2550); Tensor 27红外光谱仪 (Thermo, 美国, KBr压片); NMR用Bruker公司AVANCE-500型测定, TMS为内标; ESIMS和HRESIMS分别用美国Waters Autospec-3000质谱仪和德国Bruker公司的API QSTAR脉冲高分辨质谱仪; 高效液相色谱采用美国安捷伦GC/MS联用仪 (HP6890/5975C); ELX-800酶标仪购自美国宝特公司; 薄层色谱硅胶板、柱色谱用硅胶 (200~300目) 和硅胶H均产自青岛海洋化工厂; Sephadex LH-20凝胶为瑞士GE公司生产; 反相材料C-8为日本FU-JI公司生产, 型号为MB100-40/75, 直径40~75 μm.提取分离用石油醚、乙醇、乙酸乙酯、甲醇等试剂均为工业用化学纯品; 高效液相色谱用色谱级试剂.
3.2 真菌材料来源、菌种纯化与发酵
真菌样品于2015年6月采自海南省鹦哥岭, 由中国热带农业科学院热带生物技术研究所黄圣卓副研究员鉴定为多形炭角菌 (Xylaria polymorpha).将真菌材料用清水清洗, 用75%的酒精浸泡1 min, 无菌水冲洗3遍, 来进行表面消毒.将组织进行切片 (1 cm2左右), 放置于固体PDA培养基中, 于25 ℃恒温培养, 待菌丝长出之后进行转接纯化.分离纯化得到的菌种标本 (YGL-017) 保存于中国热带农业科学院热带生物技术研究所赵友兴研究组.菌种以固体PDA为培养基, 试管斜面, 4 ℃冷藏与冰箱中.
用大米固体培养基 (80 g大米和120 mL水, 1000 mL锥形瓶) 进行大规模发酵.首先将菌种转接到10个培养皿中培养一周, 再将转接的菌丝接种到准备好的300个装有大米培养基的锥形瓶 (1000 mL) 中, 于室温静置培养30 d左右.
3.3 提取与分离
发酵液用乙酸乙酯萃取3次, 得到浸膏45.0 g.对浸膏进行减压硅胶柱层析, 用石油醚-乙酸乙酯 (V:V=15:1~0:1, 每次3 L) 进行梯度洗脱得到10个馏分 (Fr.1~Fr.10). Fr.3 (5.6 g) 经ODS柱甲醇-水 (V:V=1:5, 2:3, 3:2, 4:1, 1:0) 进行梯度洗脱获得3个亚组分 (Fr.3-1~Fr.3-3). Fr.3-1 (0.18 g) 经半制备HPLC分离 (ODS柱, 85% MeOH/H2O) 得到化合物11 (tR 8.5 min, 3.0 mg) 和12 (tR 12.8 min, 5.0 mg). Fr.3-2 (1.7 g) 经凝胶Sephadex LH-20柱色谱分离 (CHCl3/MeOH, V:V=1:1, 800 mL), 再用硅胶柱 (石油醚/乙酸乙酯, V:V=6:1, 600 mL) 反复纯化得到化合物7 (2.2 mg) 和8 (10.0 mg). Fr.4 (4.5 g) 经反相柱RP-18 (甲醇/水, V:V=3:7~9:1, 每次1 L) 洗脱得到5个亚组分Fr.4-1~Fr.4-5. Fr.4-1经半制备HPLC分离 (ODS柱, 80% MeOH/H2O) 得到化合物13 (tR 7.0 min, 3.0 mg) 和14 (tR 14.0 min, 5.0 mg). Fr.4-3由硅胶柱 (石油醚/丙酮, V:V=5:1, 500 mL) 反复纯化得到化合物15 (2.4 mg)、16 (7.5 mg)、17 (5.7 mg). Fr.4-5先经硅胶柱石油醚/乙酸乙酯 (V:V=5:1, 1.2 L) 洗脱纯化, 再经凝胶柱Sephadex LH-20纯化 (CHCl3/MeOH, V:V=1:1, 600 mL) 得到2 (4.7 mg) 和3 (3.7 mg). Fr. 5 (3.6 g) 经硅胶柱石油醚/乙酸乙酯 (V:V=4:1) 洗脱得到2个亚组分 (Fr.5-1~Fr.5-2). Fr.5-1经半制备HPLC分离 (ODS柱, 85% MeOH/H2O) 得到化合物9 (tR 8.0 min, 20.3 mg) 和10 (tR 11.0 min, 3.2 mg). Fr.6 (6.5 g) 经反相柱RP-18 (MeOH/H2O, V:V=3:5, 1:1, 4:1, 9:1, 每梯度300 mL) 获得3个亚组分 (Fr.6-1~Fr.6-3). Fr.6-1经反复RP-18反相柱 (MeOH/H2O, V:V=3:7~9:1) 和硅胶色谱柱 (石油醚-乙酸乙酯, V:V=3:1~1:1) 依次得到18 (3.8 mg), 19 (5.3 mg), 20 (2.3 mg) 和21 (4.7 mg). Fr.8 (7.3 g) 经凝胶柱Sephadex LH-20 (CHCl3/MeOH, V:V=1:1, 2 L) 和RP-18 (MeOH/H2O, V:V=1:5, 2:3, 3:2, 4:1, 1:0, 1 L) 得到4个亚组分. Fr.8-2经硅胶柱石油醚/乙酸乙酯 (V:V=1:2) 和凝胶柱Sephadex LH-20 (CHCl3/MeOH, V:V=1:1, 600 mL) 纯化得到化合物1 (6.3 mg), 5 (2.0 mg) 和6 (3.0 mg).化合物4 (30.0 mg) 是Fr.8-2经石油醚/乙酸乙酯 (V:V=1:2~1:3, 800 mL) 分离得到.
Polymorphine A (1):白色无定型粉末, 分子式C15H24O5. m.p. 103~105 ℃; [α]D25-26.5 (c 0.02, MeOH); 1H NMR和13C NMR谱数据见表 1; IR (KBr) νmax: 3384, 2964, 2945, 1765, 1460, 1390, 1096 cm-1; ESIMS positive m/z: 307 [M+Na]+; HRESIMS calcd for C15H24O5Na [M+Na]+ 307.1516, found 307.1517.
Polymorphine B (2):白色晶体, 分子式C23H32O4.m.p. 125~128 ℃; UV-vis (MeOH) λmax [log e (L·mol-1· cm-1)]: 202 (5.27), 228 (4.43), 282 (4.09) nm; [α]D25-21.5 (c 0.01, MeOH); 1H NMR和13C NMR谱数据见表 1; IR (KBr) νmax: 2936, 1719, 1610, 1585, 1450, 1269, 1104 cm-1; ESIMS positive m/z: 395 [M+Na]+; HRESIMS calcd for C23H32O4Na [M+Na]+ 395.2193, found 395.2190.
2-甲基-2-(4-羟甲基苯基) 氧杂环戊烷 (3):淡黄色油状液体. [α]D25-3.4 (c 0.05, MeOH); 1H NMR和13C NMR谱数据见表 1; IR (KBr) νmax: 3444, 2968, 2870, 1708, 1450, 1267, 1035 cm-1; ESIMS positive m/z: 192 [M+Na]+; HRESIMS calcd for C12H16O2Na [M+Na]+ 215.1043, found 215.1045.
3.4 Polymorphine B (2) 的单晶X衍射测定
在石油醚-乙酸乙酯混合溶剂中, 溶剂挥发得到针状晶体, 采用Bruker Smart CCD衍射仪 (Mo-Kα靶, λ=0.71073 Å).斜方晶, C23H32O4, M=395.21; a=6.3399(4) Å, b=9.4636(6) Å, c=34.365(2) Å; V=2061.8(2) Å3, Dcalcd=1.200 mg·m-3, F(000)=808; β=90.00;晶体大小0.39 mm×0.26 mm×0.15 mm; T=296(2) K; 空间点群P21, Z=4, μ=0.080 mm-1; 收集衍射点数为5136;优化方法基于F2全矩阵最小二乘法; 最后修正值R1=0.0468, wR2=0.0960, R=0.0819;化合物2的CIF文件存放于英国剑桥X单晶衍射数据中心, CCDC编号为1497291, 具体信息可通过https://www.ccdc.cam.ac.uk/deposit/获取.
3.5 活性测试
3.5.3 抗线虫活性
线虫培养与线虫悬液制备:接种线虫Panagrellus redivivus到燕麦片培养基 (燕麦片20 g, 水60 mL, 混匀后分装于250 mL锥形瓶中, 121 ℃高温灭菌30 min), 于28 ℃下培养7~10 d.按贝曼漏斗法用无菌水在4层擦镜纸上过滤线虫两次, 获得线虫悬液.将1.0 mg样品用20 μL二甲基亚砜 (DMSO) 溶解制成样品溶液.在96孔板, 每孔加30 μL线虫悬液 (约200~300条线虫), 无菌水65 μL, 纯品溶液5 μL, 样品终浓度为2.5 mg/mL.空白对照为上述处理中用等量的DMSO替代样品溶液.混匀后于室温下培养24 h, 于解剖镜下观察计数线虫死亡数, 统计的线虫数量不少于100条.线虫死亡率 (%)=死亡线虫数/计数线虫总数×100%, 线虫校正死亡率 (%)=(处理线虫死亡率-对照线虫死亡率)/(1-对照线虫死亡率).
辅助材料 (Supporting Information) 新化合物1~3的谱图数据和HRESIMS.这些材料可以免费从本刊网站 (http://sioc-journal.cn/) 上下载.
3.5.2 α-葡萄糖苷酶抑制活性
使用PNPG法对化合物的α-糖苷酶抑制活性进行评价.使用pH 6.8的0.2 mol/L磷酸氢二钠-磷酸二氢钠缓冲溶液 (PBS溶液) 作为反应溶液.待测样品溶液的配制: 1 mg待测化合物溶解于20 μL的二甲基亚砜后, 取5 μL的该溶液加至45 μL的PBS溶液中, 配制成5 mg/mL的待测化合物溶液.将以下各溶液混匀于96孔酶标板后置于酶标仪中:实验组10 μL待测样品溶液+70 μL PBS溶液+20 μL PBS溶液溶解的2 U/mL α-糖苷酶溶液 (A), 阴性对照10 μL 10%的DMSO-PBS溶液+70 μL PBS溶液+20 μL PBS溶液溶解的2 U/mL α-糖苷酶溶液 (B), 阳性对照10 μL的5 mg/mL阿卡波糖溶液+70 μL PBS溶液+20 μL PBS溶液溶解的2 U/mL α-糖苷酶溶液, 背景对照10 μL待测样品溶液+90 μL PBS溶液 (A0), 空白对照10 μL 10%的DMSO-PBS溶液+90 μL的PBS溶液 (B0).将96孔板于37 ℃放置15 min后, 各组均加入2.5 mmol/L的4-硝基苯基-β-D-吡喃葡萄糖苷 (PNPG) 溶液20 μL; 将96孔板于37 ℃放置30 min后, 加入0.2 mol/L的Na2CO3终止液80 μL, 于酶标仪405 nm波长下测量每孔的OD值吸光度并计算化合物对α-糖苷酶的抑制活性.计算公式如下:抑制率 (%)=[(B-B0)-(A-A0)]/(B-B0)×100%[23]
3.5.1 AChE抑制活性
使用Ellman[22]法对化合物的乙酰胆碱酯酶抑制活性进行评价.使用pH 8.0的0.1 mol/L磷酸氢二钠-磷酸二氢钠缓冲溶液 (PBS溶液) 作为反应溶液.待测样品溶液的配制: 1 mg待测化合物溶解于20 μL的二甲基亚砜后, 取2 μL的该溶液加至98 μL的PBS溶液中, 配制成1 mg/mL的待测化合物溶液.将以下各溶液混匀于96孔酶标板后置于酶标仪中:实验组10 μL待测样品溶液+110 μL PBS溶液+40 μL PBS溶液溶解的0.1 U/mL乙酰胆碱酯酶溶液 (A), 阴性对照10 μL 2%的DMSO-PBS溶液+40 μL PBS溶液溶解的0.1 U/mL乙酰胆碱酯酶溶液 (B), 阳性对照10 μL的6.66 μmol/L他克林溶液+110 μL PBS溶液+40 μL PBS溶液溶解的0.1 U/mL乙酰胆碱酯酶溶液, 背景对照10 μL待测样品溶液+150 μL PBS溶液 (A0), 空白对照10 μL 2%的DMSO-PBS溶液+150 μL的PBS溶液 (B0) 将96孔板于30 ℃放置20 min后, 各组均加入等体积混合的6.25 mmol/L的5, 5'-二硫代双 (2-硝基苯甲酸) 溶液与6.25 mmol/L的硫代乙酰胆碱溶液40 μL; 将96孔板于30 ℃放置30 min后, 于酶标仪405 nm波长下测量每孔的OD值吸光度并计算化合物对乙酸胆碱酯酶的抑制活性.计算公式如下:抑制率 (%)=[(B-B0)-(A-A0)]/(B-B0)×100%.
-
-
[1]
Lee, I. K.; Jang, Y. W.; Kim, Y. S.; Yu, S. H.; Lee, K. J.; Park, S. M.; Oh, B. T.; Chae, J. C.; Yun, B. S. J. Antibiot. 2009, 62, 163. doi: 10.1038/ja.2008.20
-
[2]
Shiono, Y.; Motoki, S.; Koseki, T.; Murayama, T.; Tojima, M.; Kimura, K. I. Phytochemistry 2009, 70, 935. doi: 10.1016/j.phytochem.2009.03.023
-
[3]
Jang, Y. W.; Lee, I. K.; Kim, Y. S.; Lee, S. K.; Lee, H. J.; Yu, S. H.; Yun, B. S. J. Antibiot. 2007, 60, 696. doi: 10.1038/ja.2007.89
-
[4]
Shan, W.-G.; Chen, X.-X.; Ying, Y.-M.; Zhan, Z.-J. HeIv. Chim. Acta 2011, 94, 1254. doi: 10.1002/hlca.v94.7
-
[5]
William, A. A.; Latchezar, S. T. J. Nat. Prod. 1992, 55, 1454. doi: 10.1021/np50088a011
-
[6]
He, J.-B.; Tao, J.; Miao, X.-S.; Bu, W.; Zhang, S.; Dong, Z.-J.; Li, Z.-H.; Feng, T.; Liu, J.-K. Fitoterapia 2015, 102, 1. doi: 10.1016/j.fitote.2015.01.022
-
[7]
李齐激, 王冲, 李继新, 张敬杰, 刘亚华, 潘炉台, 中国药学杂志, 2014, 49, 550.Li, Q.-J.; Wang, C.; Li, J.-X.; Zhang, J.-J.; Liu, Y.-H.; Pan, L.-T. Chin. Pharm. J. 2014, 49, 550 (in Chinese).
-
[8]
裴刚, 周朴华, 何贵霞, 杜方麓, 蒋道松, 天然产物研究与开发, 2004, 16, 213. doi: 10.3969/j.issn.1001-6880.2004.03.008Pei, G.; Zhou, P.-H.; He, G.-X.; Du, F.-L.; Jiang, D.-S. Nat. Prod. Res. Dev. 2004, 16, 213 (in Chinese). doi: 10.3969/j.issn.1001-6880.2004.03.008
-
[9]
Dean, V.-J.; Herfried, G. Tetrahedron 1997, 53, 617. doi: 10.1016/S0040-4020(96)01023-X
-
[10]
黄学石, 郜嵩, 范丽华, 庾石山, 梁晓天, 中国中药杂志, 2004, 29, 1108. doi: 10.3321/j.issn:1001-5302.2004.11.028Huang, X.-S.; Gao, S.; Fan, L. H.; Yu, S.-S.; Liang, X.-T. China J. Chin. Mater. Med. 2004, 29, 1108 (in Chinese). doi: 10.3321/j.issn:1001-5302.2004.11.028
-
[11]
Zhou, W.-W.; Guo, S.-X. Chem. Nat. Compd. 2009, 45, 124. doi: 10.1007/s10600-009-9229-x
-
[12]
Li, W.-H.; Chang, S.-T.; Chang, S.-C.; Chang, H.-T. Nat. Prod. Res. 2008, 22, 1085. doi: 10.1080/14786410802267510
-
[13]
Luo, P.; Su, J.; Zhu, Y.-L.; Wei, J.-H.; Wei, W.-X.; Pan, W.-G. Nat. Prod. Res. 2016, 30, 2190. doi: 10.1080/14786419.2016.1160231
-
[14]
Hybelbauerová, S.; Sejbal, J.; DracInsky, M.; Hahnová, A.; Koutek, B. Chem. Biodiversity 2008, 5, 743. doi: 10.1002/(ISSN)1612-1880
-
[15]
Liu, X.-H.; Miao, F.-P.; Liang, X.-R.; Ji, N.-Y. Nat. Prod. Res. 2014, 28, 1182. doi: 10.1080/14786419.2014.923996
-
[16]
Kwon, H. C.; Zee, S. D.; Cho, S. Y.; Choi, S. U.; Lee, K. R. Arch. Pharm. Res. 2002, 25, 851. doi: 10.1007/BF02977003
-
[17]
Gao, H.; Hong, K.; Zhang, X.; Liu, H.-W.; Wang, N.-L.; Zhuang, L.; Yao, X.-S. HeIv. Chim. Acta 2007, 90, 1165. doi: 10.1002/(ISSN)1522-2675
-
[18]
Fangkrathok, N.; Sripanidkulchai, B.; Umehara, K.; Noguchi, H. Nat. Prod. Res. 2012, 27, 1611.
-
[19]
Amagata, T.; Tanaka, M.; Yamada, T.; Doi, M.; Minoura, K.; Ohishi, H.; Yamori, T.; Numata, A. J. Nat. Prod. 2007, 70, 1731. doi: 10.1021/np070165m
-
[20]
鄂恒超, 周巍, 刘宝姝, 汤华, 孙鹏, 李玲, 张文, 中国海洋药物, 2013, 32, 8. http://mall.cnki.net/magazine/Article/HYYW201306002.htmE, H.-C.; Zhou, W.; Liu, B.-S.; Tang, H.; Sun, P.; Li, L.; Zhang, W. Chin. J. Marine Drugs 2013, 32, 8 (in Chinese). http://mall.cnki.net/magazine/Article/HYYW201306002.htm
-
[21]
Butova, E. D.; Barabash, A. V.; Petrova, A. A.; Kleiner, C. M.; Schreiner, P. R.; Fokin, A. A. J. Org. Chem. 2010, 75, 6229. doi: 10.1021/jo101330p
-
[22]
Ellman, G. L.; Courtney, K. D.; Jr., V. A.; Featherstone, R. M. Biochem. Pharmacol. 1961, 7, 88. doi: 10.1016/0006-2952(61)90145-9
-
[23]
李婷, 张小东, 宋聿文, 刘建文, 中国临床药理学与治疗学, 2005, 10, 1128. doi: 10.3969/j.issn.1009-2501.2005.10.011Li, T.; Zhang, X.-D.; Song, Y.-W.; Liu, J.-W. Chin. J. Clin. Pharmacol. Ther. 2005, 10, 1128 (in Chinese). doi: 10.3969/j.issn.1009-2501.2005.10.011
-
[1]
-

图 2 化合物1~3的主要COSY和HMBC相关信号
Figure 2 Key 1H-1H COSY and HMBC correlations of compounds 1~3
表 1 化合物1~3的NMR数据
Table 1. NMR data of 1~3 (1 in CD3OD, 2 and 3 in CDCl3)
No. 1 2 3 δC δH (J in Hz) δC δH (J in Hz) δC δH (J in Hz) 1 30.0 (t) 2.09, m 1.48, m 85.1 (d) 4.90, dd (4.3, 11.5) 2 26.5 (t) 1.76, m 1.72, m 24.5 (t) 1.78, m 1.74, m 84.3 (s) 3 78.5 (d) 3.24, dd (6.2, 10.0) 39.7 (t) 1.52, m 1.50, m 39.6 (t) 2.19, m 2.02, m 4 39.3 (s) 33.1 (s) 25.9 (t) 1.99, m 1.80, m 5 45.9 (d) 1.61, dd (3.0, 13.0) 51.4 (d) 1.02, dd (3.4, 11.5) 67.7 (t) 4.01, m 3.90, m 6 17.4 (t) 1.66, m 1.62, m 17.7 (t) 1.50, m 1.42, m 29.8 (q) 1.52, s 7 28.2 (t) 2.00, m 1.98, m 24.1 (t) 1.64, m 1.60, m 147.9 (s) 8 78.8 (s) 34.7 (d) 2.56, m 127.1 (d) 7.32, d (8.0) 9 79.6 (s) 57.4 (d) 1.91, d (8.0) 125.1 (d) 7.37, d (8.0) 10 39.1 (s) 39.1 (s) 139.0 (s) 11 174.4 (s) 108.4 (d) 5.03, s 125.1 (d) 7.37, d (8.0) 12 78.4 (t) 4.52, d (8.5) 3.95, d (8.5) 71.7 (t) 3.91, dd (8.3, 9.0) 3.53, dd (8.3, 9.0) 127.1 (d) 7.32, d (8.0) 13 15.4 (q) 0.85, s 12.4 (q) 1.20, s 65.3 (t) 4.67, s 14 17.6 (q) 1.26, s 33.0 (q) 0.92, s 15 28.5 (q) 1.04, s 22.3 (q) 0.92, s 16 54.1 (q) 2.69, s 17 165.8 (s) 18 130.8 (s) 19/23 129.7 (d) 8.05, d (8.0) 20/22 128.5 (d) 7.44, t (7.9) 21 133.0 (d) 7.55, t (7.4)  下载: 导出CSV
下载: 导出CSV
-





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 15
- 文章访问数: 2326
- HTML全文浏览量: 424
 下载:
下载:
