Citation:
Yi Wang, Runze Liu, Ming Shi, Panwang Zhou, Keli Han, Can Li, Rengui Li. Photo-induced carbon dioxide reduction on hexagonal tungsten oxide via an oxygen vacancies-involved process[J]. Chinese Chemical Letters,
2023, 34(1): 107200.
doi:
10.1016/j.cclet.2022.02.006
Photo-induced carbon dioxide reduction on hexagonal tungsten oxide via an oxygen vacancies-involved process
English
Photo-induced carbon dioxide reduction on hexagonal tungsten oxide via an oxygen vacancies-involved process
State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian National Laboratory for Clean Energy, Dalian 116023, China
b.
University of Chinese Academy of Sciences, Beijing 100049, China
c.
Institute of Molecular Sciences and Engineering, Shandong University, Qingdao 266235, China
d.
State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
Received Date:
10 December 2021 Accepted Date:
03 February 2022 Revised Date:
09 January 2022 Available Online:
15 January 2023
Abstract:
Although converting the greenhouse gasses carbon dioxide (CO2) into solar fuels is regarded as a convenient means of solar energy storage, the intrinsic mechanism on how the high chemical inertness linear CO2 molecules is activated and converted on a semiconductor oxide is still elusive. Herein, by creating the oxygen vacancies on the typical hexagonal tungsten oxide (WO3), we realize the continuous photo-induced CO2 reduction to selectively produce CO under light irradiation, which was verified by isotope labeling experiment. Detailed oxygen vacancies evolution investigation indicates that light irradiation can simultaneously induce the in-situ formation of oxygen vacancies on hexagonal WO3, and the oxygen vacancies promote the adsorption and activation of CO2 molecules, leading to the CO2 reduction to CO on the hexagonal WO3via an oxygen vacancies-involved process. Besides, the existence of water further promotes the formation of CO2 reduction intermediate, further promote the CO2 photoreduction. Our work provides insight on the mechanism for converting CO2 into CO under light irradiation.
Converting greenhouse gasses CO2 in atmosphere into value-added chemical products holds promise to address the current and future demand of energy supply and environmental disasters [1, 2]. Although photo-induced CO2 reduction on a semiconductor is appealing and attractive [3-5], the difficulties in thermodynamics and kinetics make its process complicated and challenging. In particular, the adsorption and activation of high chemical inertness of linear CO2 on the surface of semiconductor is very difficult [6]. Oxygen vacancies are the most prevalent and widely studied anion defects with a relatively lower formation energy on heterogeneous oxide surfaces [7-11]. Normally, oxygen vacancies on the catalyst surfaces can effectively modulate both the coordination structures and electronic states of reactive molecules, thus influencing the reaction kinetics and further altering the reaction mechanisms [12-14]. However, an in-depth scenario depicting the interactions between oxygen vacancies and reactive molecular in complicated reactions is still unclear, particularly for the challenging CO2 reduction reactions [15]. Oxygen vacancies can favor the CO2 binding and subsequent CO2 activation, as well as the final dissociation to promote the catalytic process [16-18]. On the other hand, oxygen vacancies have extensively recognized to serve as the activation sites for the thermodynamically stable molecules such as CO2 and N2 [19-22]. However, in a two-step thermochemical CO2 reduction process, oxygen vacancies created by endothermic reduction of metal oxides were found to induce the effective dissociation of CO2 molecules acting as Lewis basic sites [23]. Although oxygen vacancies can facilitate the CO2 reduction in numerous reactions, intrinsic mechanism on how the high chemical inertness of linear CO2 molecules are activated and converted is inconclusive and has not fully elucidated.
It is accepted that the CO2 adsorption and activation on a heterogeneous photocatalyst involves the formation of a partially charged species CO2δ− to lower the activation barrier, in which process electron transfer from surface atoms to CO2 molecules is dominated [24]. Subsequent reduction of CO2•‒ into carbon-containing products involves the electron and proton transfer and breaking C=O bonds [25, 26]. The complex chemical environment of the photocatalyst surfaces leads to the formation of diverse intermediates, further causing complicated reaction pathways [27]. Several possible reaction pathways were proposed such as the formaldehyde pathway [28], the carbene pathway [29], but the mechanism from CO2 adsorption to catalytic reaction is still unclear. Furthermore, utilizing water as the electron donor is the ultimate solution of CO2 photoreduction, but the oxygen evolution half-reaction involves complicated four-electron transfer process and O=O bond formation, which makes the CO2 reduction process coupling with water oxidation more difficult.
In this work, taking hexagonal tungsten oxide (WO3), a semiconductor photocatalyst with unfavorable band structure as a prototype, we experimentally demonstrate the continuous CO2 reduction to selectively produce CO under light irradiation can be achieved on the hexagonal WO3. Investigations indicate that light irradiation induces the in-situ formation of oxygen vacancies on the hexagonal WO3, and the oxygen vacancies promote the absorption and activation of CO2 molecules, thus making the photo-induced CO2 reduction takes place via an oxygen vacancies-involved process.
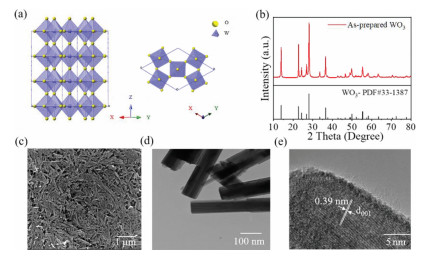
Hexagonal tungsten trioxide (WO3) has been intensively studied as a candidate photocatalyst thanks to its advantages in light harvesting properties and chemical stabilities over a wide range of acidity [7-10, 12, 30]. It is widely-held that the conduction band edge of hexagonal WO3 locates at ~0.2 eV vs. RHE, which is more positive than CO2 reduction to carbon-containing products energy levels, namely, its conduction band is thermodynamically unfavorable to drive the direct reduction of CO2 molecules. Analyzing from the microstructure, hexagonal WO3 is composed of WO6 octahedral unit cells which share its corner oxygen atom to construct a defect perovskite-like ReO3 structure (Fig. 1a). The intrinsic structure of hexagonal WO3 leads to incidental oxygen atom leakage, creating oxygen vacancies. Correspondingly, the decrease ratio of structural W=O double bonds in the unit cell to single bonds is proposed to correlate the oxygen vacancies generation process [31]. Chromic properties result from the color centers is common in the WO3 material, generally being attributed to electron excess localized on W ions (W5+), which is closely related to the oxygen vacancies [32].
Figure 1
Figure 1.
Schematic structure and characterization of as-prepared hexagonal WO3. (a) Scheme illustration of hexagonal WO3 crystal structure. (b) XRD pattern of as-prepared WO3. (c) SEM image of hexagonal WO3. (d, e) High-resolution TEM images of hexagonal WO3.
The hexagonal WO3 was employed in this work and prepared via a hydrothermal process as reported [33]. Detailed synthesis, characterization and CO2 photoreduction process was shown in Supporting information. All the XRD diffraction peaks of as-synthesized sample can be well indexed to hexagonal phase WO3 with no characteristic peak of sub stoichiometric WO3 or hydrates was observed (Fig. 1b). The morphology of the hexagonal WO3 exhibits a rod-like shape with diameter of ~50 nm and rod length of 1–2 µm (Figs. 1c and d, Figs. S1 and S2 in Supporting information). A regular lattice fringe spacing about 0.39 nm was shown in high-resolution TEM image, which can be attributed to the representative (001) plane of the hexagonal WO3, demonstrating that the prepared WO3 nanorod grew along the [001] crystal direction (Fig. 1e). Mott-Schottky analysis was applied to determine the conduction band edge of as-prepared WO3 (Fig. S3 in Supporting information). As an n-type semiconductor, the conduction band edge of hexagonal WO3 was estimated to be 0.07 eV, similar to the results previously reported [34]. The band gap (Eg) was calculated to be 2.68 eV (Figs. S4 and S5 in Supporting information).
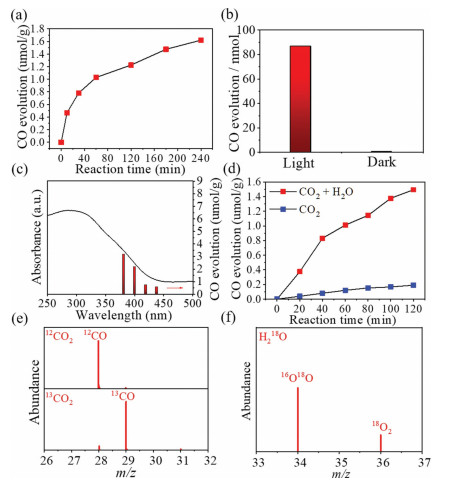
Photo-induced CO2 reduction on the hexagonal WO3 was examined under CO2 atmosphere with the presence of water vapor using Xe lamp as light source. As shown in Fig. 2a, surprisingly, we observed that the continuous producing of CO gas can be obviously detected under light irradiation, which means that CO2 can be reduced to CO under this condition. As contrast, no CO product was detected in the same condition without light irradiation, illustrating that the CO2 reduction should be responsive to the light-induced photoreduction process (Fig. 2b). Furthermore, we verified that either no CO was detected in the case when the hexagonal WO3 was absence (Fig. S6 in Supporting information). Structural evolution under the illumination condition was further confirmed by Raman spectroscopy (Fig. S7 in Supporting information). Moreover, CO2 reduction performance was also examined under different wavelength ranges. As depicted in Fig. 2c, CO2 photoreduction towards CO is significantly correlated with the light absorption ranges of hexagonal WO3, confirming that the CO2 reduction is associated with the photo excitation of hexagonal WO3 catalyst and the subsequent charge-involved photoreduction process. Additionally, comparative experiments with or without introducing H2O vapor were conducted. As can be seen from Fig. 2d, a remarkable difference of the performance was clearly observed under the cases with or without H2O. When the H2O vapor was introduced into the reactor, the photoreduction activity was more than an order of magnitude of that without H2O, indicating that H2O molecules exactly participate in the CO2 photoreduction process.
Figure 2
Figure 2.
(a) Photocatalytic activities of CO2 reduction on hexagonal WO3. (b) Control experiments of CO2 reduction under light and dark conditions. (c) CO2 photoreduction to CO on hexagonal WO3 under different wavelengths. (d) The comparison of CO2 photoreduction with or without introducing H2O vapor. (e, f) Mass spectra extracted from GC–MS analysis of product with isotope labeling using 13CO2 and H218O.
Besides, the photocatalytic activity of CO2 reduction in gas and liquid phase were also estimated. Under the same irradiation condition, the photocatalytic activity in gas phase is slightly higher than that in liquid phase, which may due to the weak solubility of CO2 (Fig. S8 in Supporting information).
To certify that the reliable product comes from CO2 photoreduction rather than other contaminants, isotope labeling experiment was performed using 13CO2 as the reactant, while the product was analyzed by GC–MS. As shown in Fig. 2e, for non-labeled CO2, molecular ion peak at m/z = 28 indicates that the product is CO, while replacing the reactant by 13CO2, molecular ion peak at m/z = 29 evidently showed up without any other products, indicating the selective formation of 13CO, giving clear evidence that the produced CO is originated from the CO2 input with a high selectivity. To verify the attendance of water, control experiment with 18O labeling H2O was also examined. In the mass spectrum shown in Fig. 2f, two peaks appeared at m/z values of 34 and 36, which can be attributed to the ionic 16O18O and 18O2, respectively. The detected O2 undoubtedly confirms that the water oxidation takes place during CO2 photoreduction. Therefore, we can come to a conclusion that the CO2 photoreduction can be realized on the hexagonal WO3. It should be noted that although H2O molecules were confirmed to participate in the reaction process, no detectable H2 was observed in the experiment, which may be due to fact that the hydrogen species formed in the reaction adsorb on the surface of the catalyst. To investigate the phenomenon, examination of H2 adsorption was conducted for the hexagonal WO3 with or without light treatment (Fig. S9 in Supporting information). As expected, we found that the hexagonal WO3 after photocatalytic reaction under light irradiation shows a drastically enhanced H2 adsorption capability compared to the fresh WO3, which is proposed to be the reason why no H2 but only CO was detected in the experiment.
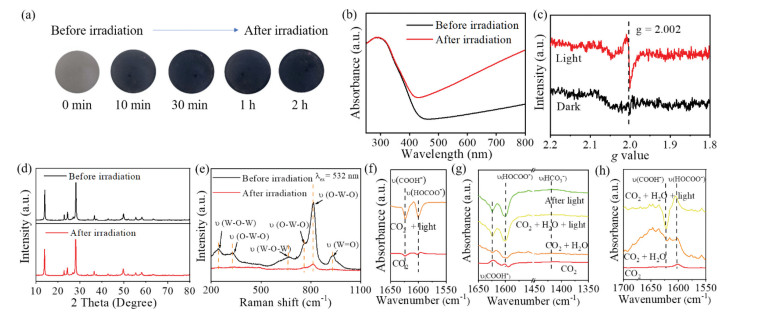
The results above evidently demonstrate the occurrence of photo-induced CO2 reduction to CO on the hexagonal WO3, however, how the reaction takes place is still unclear. Considering the structure of hexagonal WO3, each oxygen atom bounds to two W atoms in the lattice, whereas there may be missing oxygen atoms at the boundary or corner sites on the surface. In addition, the photogenerated holes with high oxidation potential may oxidize the lattice oxygen of the hexagonal WO3 and create oxygen vacancies on the surface, which attends the CO2 reduction and activation subsequently. As depicted in Fig. 3a, when the hexagonal WO3 was irradiated under light, the apparent color gradually changes from light gray to dark blue with irradiation time prolonging. UV–vis diffusion reflection spectroscopy was then employed to investigate the absorbance during this color-changing process (Fig. 3b). Obviously, the absorption in the region of 500–800 nm dramatically increases, while the absorption at the band edge keeps nearly constant. In general, the light absorption increase in visible light region for semiconductors can be attributed to the formation of low-valued metal species [35]. Here, the increase in light absorption can be attributed to the generation of pentavalent tungsten (W5+), which also correlates the generation of oxygen vacancies during the CO2 photoreduction reaction on the hexagonal WO3. Electron spin resonance (ESR) spectra were therefore employed to further verify the formation of oxygen vacancies. As depicted in Fig. 3c, no significant signal was observed in dark condition, whereas an obvious signal at g = 2.002 appears once light irradiated, which clearly evidenced the formation of single-electron-trapped oxygen vacancies. Furthermore, the hexagonal WO3 samples before and after irradiation exhibit comparable XRD diffraction peaks and intensities (Fig. 3d), indicating the maintained crystallinity of the hexagonal WO3 with light-induced formation of oxygen vacancies, which was also supported by the unchanged lattice spacing of the hexagonal WO3 compared with the fresh ones (Fig. S10 in Supporting information). Fig. 3e depicts the Raman spectra of the hexagonal WO3 before and after light irradiation. The bands with Raman shift at 760 cm−1 and 817 cm−1 can be ascribed to O-W-O stretching mode, whilst peaks at 250 and 324 cm−1 belong to the stretching mode of W-O-W [36]. The Raman peak at 940 cm−1 can be assigned to the W=O double bond stretching mode located at the boundaries. After light irradiation to generate oxygen vacancies, the intensity of all characteristic peaks dramatically decreases. Besides, the defects of nanocrystals can be reflected on the width of Raman shift [37]. The peak width at half-height at around 806 cm−1 decreased obviously after irradiation, which supports the formation of oxygen vacancies laterally.
Figure 3
Figure 3.
(a) Photographs of the hexagonal WO3 under light irradiation. (b) UV–visible spectra of the hexagonal WO3 before and after light irradiation. (c) In-situ EPR spectra of the hexagonal WO3 before and after light irradiation. (d) XRD pattern of the hexagonal WO3 before and after the photocatalytic reaction. (e) In-situ Raman spectra of the hexagonal WO3 before and after light irradiation. In-situ FTIR spectra of (f) CO2 and (g) CO2 and water vapor interaction with fresh WO3 in the dark and under photoirradiation subsequently. (h) CO2 and water vapor interaction with pre-irradiated WO3 in the dark and under photoirradiation subsequently.
To further build a connection between the oxygen vacancies formation and CO2 photoreduction and provide an unambiguous interpretation of the water function of in the reaction process, in-situ transient Fourier transform infrared spectroscopy (FTIR) experiment were conducted (Figs. 3f-h). The spectra were obtained by subtracting WO3 background in Ar atmosphere. When sole CO2 was introduced to the reaction system, no obvious weak was shown in Fig. 3f, which can be attributed to the weak absorption of CO2. When WO3 was under irradiation, two negative peaks corresponding to COOH− and HOCOO− were shown, which may due to the reduction of chemical absorbed CO2 [38]. As for the situation that CO2 purged through the water (Fig. 3g), a broaden peak at around 1650 cm−1 arose, which belongs to the –OH, besides, a weak peak at 1425 cm−1 assigned to HCO3− occurred, which indicate that attendance of water promotes the formation of HCO3− intermediate. Besides, under the same irradiation condition, the peaks corresponding to COOH− and HOCOO− decrease with a larger magnitude, which support the inference above. It is noticeable that after irradiation the peak around 1425 cm−1 reoccurred, suggesting that HCO3− can be formed with the help of water. In addition, the formation and decomposition of the HCO3− keeps dynamic balance during the irradiation.
To figure out the function of oxygen vacancies, the catalyst was pre-irradiated in Ar to induce oxygen vacancies formation, followed by the same experiment steps as above (Fig. 3h). The spectra were normalized according to the transmittance. It is noticeable that when the catalyst was kept in the dark state under humid CO2 atmosphere, no peak corresponding to CO2 reaction intermediate was found, which imply that photo-generated electrons and holes are indispensable. Different from the fresh catalyst, the spectrum of the catalyst containing oxygen vacancies shows a small peak corresponding to HOCOO− species after CO2 adsorption. A plausible explanation for is that one oxygen atom of CO2 fills the oxygen vacancy and the other oxygen atom bind with hydroxy group on the surface to form a bending structure [39]. Compared with the spectrum of intrinsic WO3 under same condition, a larger hydroxyl peak was shown. During the irradiation, the peak belongs to COOH− decreases but is different from that HOCOO− species. With the help of H2O molecules, the oxygen vacancies facilitate the formation of HOCOO−, namely HCO3− intermediate and consequently promote CO2 reduction.
The aforementioned results proved the creation of oxygen vacancies via the light-induced process during photocatalytic reactions on the hexagonal WO3 while the photogenerated electrons will reduce W6+ to W5+. It should be emphasized that such oxygen vacancies generation process is reversible when the catalyst was exposed to O2-containing atmosphere. The created oxygen vacancies on the surface of hexagonal WO3 can be eliminated by O2 (Figs. S11 and S12 in Supporting information), and the oxygen vacancies can be recovered again after irradiated by light, completing the oxygen vacancies-containing chemical redox loop on the surface. The recyclable CO2 photoreduction during this reversible oxygen vacancies-containing process were also confirmed, further confirming the indispensable role of oxygen vacancies in this process (Fig. S13 in Supporting information). It can be therefore concluded that irradiation of WO3 created the oxygen vacancies on the surface, which serves as the activation site of CO2 reduction. The existence of light-induced oxygen vacancies makes it possible for hexagonal WO3 to drive CO2 reduction, whereas an appropriate amount of oxygen vacancies is needed. To further verify the essential role oxygen vacancies on the photo-induced CO2 reduction on the hexagonal WO3, we synthesized a series of oxygen vacancies-containing samples via treating the hexagonal WO3 under the atmosphere of H2 gas (Fig. S14 in Supporting information). The reduction of WO3 to produce oxygen vacancies were confirmed by the UV–vis spectra of the samples treated under different conditions. The photocatalytic activities increase obviously with increasing the content of oxygen vacancies to reach the maximum. Excess amount of the oxygen vacancies may destroy the structure of hexagonal WO3 and result in a decline in photocatalytic activities.
DFT calculation was also utilized to verify the function of oxygen vacancies. We first simulated the most possible oxygen vacancy sites on the hexagonal WO3 surface. As shown in Fig. S15a (Supporting information), the comparison of three modes of oxygen vacancy were presented on the representative (100) surface. The relative defect formation energy was calculated during removing an oxygen atom from the corresponding location, the structure of Type Ⅲ with lowest defect formation energy was found to be the most stable oxygen vacancy site. The adsorption mode of CO2 molecules on the perfect (100) and defective (100) surfaces was then calculated (Figs. S15b-e in Supporting information). When CO2 molecule adsorbs on a perfect (100) surface (Figs. S15b and d), the adsorption energy is −0.37 eV. Compared with the C=O in CO2 molecule, the C=O in adsorbed CO2 is not significantly elongated compared with the non-absorbed case (1.25 Å vs. 1.17 Å). In addition, the C atom in CO2 molecule forms a weak link with the surface oxygen atom, causing a bending configuration with a 136° carbon-oxygen double bond angle. In contrast, the adsorption energy of CO2 molecule adsorbed on the oxygen vacancy-containing (100) surface is −2.09 eV, which is much larger in magnitude than that on the perfect (100) surface (Figs. S15c and e). The results illustrate that CO2 molecules can be easily adsorbed on the oxygen vacancy sites. Moreover, the C=O bond in absorbed CO2 is obviously elongated from 1.17 Å to 1.457 Å and the bond angle of C=O was reduced to 124.8°. It is acceptable that the O atom in CO2 adsorbed onto the oxygen vacancy promotes the electron transfer from low-value W5+ to the O sites, leading to the elongation of C=O bond, which further intensifies the electron cloud mismatching between C and O atoms. The elongated C=O bond at this scale is much easier to break, signifying that C=O is weakened by the presence of oxygen vacancies, which effectively promotes the activation of CO2 molecules.
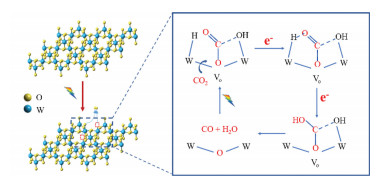
Based on the results above, oxygen vacancies can be induced on the surface of hexagonal WO3 under irradiation, which serves as the activation sites of CO2. When CO2 and water were introduced with light irradiation on WO3, a plausible reaction mechanism involved the formation of oxygen vacancies and intermediate on the hexagonal WO3 photocatalyst was proposed (Scheme 1). Initially, oxygen vacancies were induced to generate on the surface of the hexagonal WO3 by the irradiation of light. Then, oxygen atom in CO2 molecule adsorbed on the light-induced oxygen vacancies of the hexagonal WO3 surface to form bended *CO2, following by protonated by –OH adsorbed on the adjacent W atom to form adsorbed HCO3− and HCOO− intermediate. Differ from the mechanism previously reported on hydrated WO3·0.33H2O [29], since the attendance of water will lead to the formation of adsorbed H, the H species will form a hydrogen bond with the O atom in CO2. Combining with the electron transfer from the adjacent W5+, the breaking of C-O band induces filling an oxygen vacancy and formation of CO and H2O. The following irradiation will regenerate the oxygen vacancies and drive the successive reaction. The existence of photo-induced oxygen vacancies is a double-edged sword, since the presence of a small amount of oxygen vacancies will promote CO2 activation, but a large amount of that will destroy the surface structure.
Scheme 1
Scheme 1.
Schematic illustration of the proposed mechanism for photo-induced CO2 reduction on the hexagonal WO3.
In summary, we experimentally realize the photo-induced CO2 reduction to CO on the hexagonal WO3via an oxygen vacancies-involved process. Isotope labeling experiments verified the formation of oxygen with the attendance of water and lattice oxygen. The photo-induced formation of oxygen vacancies was demonstrated to closely correlate the activities of CO2 photoreduction. Combining with the in-situ FTIR, a mechanism involved oxygen vacancy was proposed that with the assistance of adsorbed water, CO2 adsorbed on oxygen vacancy was protonated to HCO3− intermediate and finally reduce to CO. Our work provides insight on the mechanism for carbon dioxide reduction on oxygen vacancy sites.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was performed by the Fundamental Research Center of Artificial Photosynthesis (FReCAP), and supported by National Natural Science Foundation of China (No. 22088102). This work was also supported by National Natural Science Foundation of China (No. 22090033). R. Li would like to thank the support from Youth Innovation Promotion Association of Chinese Academy of Sciences and Dalian Institute of Chemical Physics, CAS.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.02.006.
[1]
S. Navarro-Jaén, M. Virginie, M. Robert, et al., Nat. Rev. Chem. 5 (2021) 564–579. doi: 10.1038/s41570-021-00289-y
S.M. Sun, W. Motonori, J. Wu, et al., J. Am. Chem. Soc. 140 (2018) 6474–6482. doi: 10.1021/jacs.8b03316
[39]
Y.F. Ji, F. Luo, J. Am. Chem. Soc. 138 (2016) 15896–15902. doi: 10.1021/jacs.6b05695
Figure 1
Schematic structure and characterization of as-prepared hexagonal WO3. (a) Scheme illustration of hexagonal WO3 crystal structure. (b) XRD pattern of as-prepared WO3. (c) SEM image of hexagonal WO3. (d, e) High-resolution TEM images of hexagonal WO3.
Figure 2
(a) Photocatalytic activities of CO2 reduction on hexagonal WO3. (b) Control experiments of CO2 reduction under light and dark conditions. (c) CO2 photoreduction to CO on hexagonal WO3 under different wavelengths. (d) The comparison of CO2 photoreduction with or without introducing H2O vapor. (e, f) Mass spectra extracted from GC–MS analysis of product with isotope labeling using 13CO2 and H218O.
Figure 3
(a) Photographs of the hexagonal WO3 under light irradiation. (b) UV–visible spectra of the hexagonal WO3 before and after light irradiation. (c) In-situ EPR spectra of the hexagonal WO3 before and after light irradiation. (d) XRD pattern of the hexagonal WO3 before and after the photocatalytic reaction. (e) In-situ Raman spectra of the hexagonal WO3 before and after light irradiation. In-situ FTIR spectra of (f) CO2 and (g) CO2 and water vapor interaction with fresh WO3 in the dark and under photoirradiation subsequently. (h) CO2 and water vapor interaction with pre-irradiated WO3 in the dark and under photoirradiation subsequently.

 DownLoad:
DownLoad:

 下载:
下载:





