表 1
反应条件的优化a
Table 1.
Optimization of reaction conditions

有机膦的各类衍生物是生命体系的重要基础, 许多的芳基膦化合物已经被发现具有一定的生物活性, 因此有机膦化合物在药物化学、核酸化学中被大量地研究[1].另外, 芳基膦化合物在有机合成中又是非常重要的配体, 广泛地应用于小分子催化和过渡金属催化的反应中[2].同时这类化合物又具有特殊的光学活性, 在材料化学中也具有广泛地应用价值[3].因此发展简便、高效的芳基膦化合物的合成方法已经引起科学家们的极大关注.
2, 2'-双(二苯膦基)-1, 1'-联萘(BINAP)骨架类型的手性膦配体已被广泛应用到过渡金属催化的不对称反应中[4], 而其C—P键的构建往往需要较高活性的金属试剂(例如格氏试剂和金属锂试剂)来实现[5].因此, 这大大地限制了这种方法的实用性.近些年来, 过渡金属催化的偶联反应(Suzuki, Negishi, Stille和Heck等[6])来构建C—X键的方法已被应用到一些天然化合物及药物分子的合成中.其中过渡金属催化的构建C—P键的反应, 因其温和、高效等特点而备受关注, 钯[7]、铜[8]、镍[9]、锰[10]及银[11]催化的反应相继被报道出来.当使用Pd作催化剂时, 反应可以在非常温和的条件下实现活泼卤代烃的C—P键偶联.但对于BINAP骨架类的化合物, 由于其空间位阻较大, 产物的配位能力较强, 反应往往需要较高的催化剂负载量和较长的反应时间[12].
在此基础上, 我们通过改变溶剂、配体来调控底物与催化剂的配位能力, 高效地实现了BINAP与多种膦试剂的C—P键偶联(Eq. 1).
考虑到目标产物与催化剂有一定的配位能力, 往往需要加大催化剂的用量才可实现该类底物的反应.因此, 我们希望首先通过调控溶剂的配位能力来实现该反应的顺利进行.首先选择1a和2a作底物, 用Pd(OAc)2作催化剂, 1, 3-双(二苯基膦基)丙烷(dppp)为配体, Na2CO3作碱, 用弱配位的CH3CN作溶剂, 但目标产物收率只有9%(表 1, Entry 1).选用配位能力较弱的1, 4-二氧六环作溶剂, 反应并没有发生(表 1, Entry 2).因此用配位能力较强的二甲基亚砜(DMSO)作溶剂, 收率提高到47%(表 1, Entry 3).令人惊喜的是, 当换用N, N-二甲基甲酰胺(DMF)作溶剂时, 收率竟高达95%(表 1, Entry 4).在对配体的筛选中, 首先换用单膦配体PPh3, 反应没有发生(表 1, Entry 5).用双膦配体1, 2-双(二苯基膦基)乙烷(dppe)和1, 4-双(二苯基膦基)丁烷(dppb), 产物收率分别只有11% (表 1, Entry 6)和24%(表 1, Entry 7), 当使用dppp时, 3 h反应就完成了, 产物收率高达95% (表 1, Entry 8).在对碱的筛选中, 当换用碱性更强的碱K2CO3(表 1, Entry 9)和Cs2CO3(表 1, Entry 10), 没有产物生成, 用较弱的碱CH3COONa, 收率只有16%(表 1, Entry 11), 而使用三乙胺时, 目标产物的收率也只有15%(表 1, Entry 12), 整体上都不如Na2CO3作碱反应效果好.
|
|
(1) |
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | Solvent | Ligand | Base | Yieldb/% |
| 1 | CH3CN | dppp | Na2CO3 | 9 |
| 2 | Dioxane | dppp | Na2CO3 | Trace |
| 3 | DMSO | dppp | Na2CO3 | 47 |
| 4 | DMF | dppp | Na2CO3 | 95 |
| 5 | DMF | PPh3 | Na2CO3 | Trace |
| 6 | DMF | dppe | Na2CO3 | 11 |
| 7 | DMF | dppb | Na2CO3 | 24 |
| 8c | DMF | dppp | Na2CO3 | 95 |
| 9 | DMF | dppp | K2CO3 | Trace |
| 10 | DMF | dppp | Cs2CO3 | Trace |
| 11 | DMF | dppp | CH3COONa | 16 |
| 12 | DMF | dppp | Et3N | 15 |
| aReaction conditions: under N2, 1a (0.25 mmol), 2a (0.75 mmol), Pd(OAc)2 (5 mol%), ligand (10 mol%), base (0.75 mmol), solvent (2.0 mL), temperature is 100 ℃; reaction time is 12 h. b Isolated yield. c Reaction time is 3 h. dppp: 1, 3-bis(diphenylphosphino)propane, dppe: 1, 2-bis(diphenylphosphino)ethane, dppb: 1, 4-bis(diphenylphosphino)butane. | ||||
在进一步对催化剂的优化中, 我们首先换用了Pd(CH3CN)2Cl2作催化剂, 尽管收率也高达92%, 但反应时间需要4 h(表 2, Entry 1).随后使用了PdCl2作催化剂, 反应需要延长到12 h才能取得令人满意的收率(表 2, Entry 2).但当换用Pd2(dba)3作催化剂时, 收率降至18%(表 2, Entry 3).接着将反应温度降到80 ℃时, 反应需要15 h才能结束, 收率也高达94%(表 2, Entry 4).进一步将温度降低到50 ℃时, 该反应就不再进行了(表 2, Entry 5).随后将温度升高到了120 ℃, 反应时间缩短至2 h, 收率也高达95%(表 2, Entry 6).最后, 对催化剂的用量进行了优化.将催化剂量降低到1 mol%时, 虽然需要将反应时间延长到8 h, 但产物收率同样高达95% (表 2, Entry 7).而将催化剂量进一步降低到0.5 mol%时(表 2, Entry 8), 反应就不再进行了.
下载:
导出CSV
 |
||||
| Entry | Cat. | T/℃ | t/h | Yieldb/% |
| 1 | Pd(CH3CN)2Cl2 | 100 | 4 | 92 |
| 2 | PdCl2 | 100 | 12 | 95 |
| 3 | Pd2(dba)3 | 100 | 12 | 18 |
| 4 | Pd(OAc)2 | 80 | 15 | 94 |
| 5 | Pd(OAc)2 | 50 | 12 | Trace |
| 6 | Pd(OAc)2 | 120 | 2 | 95 |
| 7c | Pd(OAc)2 | 100 | 8 | 95 |
| 8d | Pd(OAc)2 | 100 | 12 | Trace |
| aReaction conditions: under N2, 1a (0.25 mmol), 2a (0.75 mmol), Cat. (5 mol%), dppp (10 mol%), Na2CO3 (0.75 mmol), DMF (2.0 mL). bIsolated yield. cThe amount of catalyst is 1 mol%. dThe amount of catalyst is 0.5 mol%. | ||||
基于最优条件, 我们对反应底物的普适性进行了研究(表 3).对于位阻较小的亚磷酸甲酯, 该方法也有非常好的效果, 收率高达96%(表 3, Entry 2).而对于位阻较大的二苯基膦氧底物, 温度需要升高到120 ℃, 反应才能进行, 收率可达81%(表 3, Entry 3).同时发现苯环上取代基的电性对该反应的收率影响并不是很大.对甲基取代、3, 5-二甲基取代和对甲氧基取代的底物, 收率分别为73%, 85%和76%(表 3, Entries 4~6).而对位上含有吸电子基氟的底物, 收率也高达72%(表 3, Entry 7).另外我们还尝试了将取代基苯换成了萘, 反应也取得了良好的效果, 收率为70%(表 3, Entry 8).
下载:
导出CSV
 |
|||
| Entry | 3 | R | Yieldb/% |
| 1 | 3a | OEt | 95 |
| 2 | 3b | OMe | 96 |
| 3 | 3c | C6H5 | 81 |
| 4 | 3d | 4-MeC6H4 | 73 |
| 5 | 3e | 3, 5-Me2C6H3 | 85 |
| 6 | 3f | 4-MeOC6H4 | 76 |
| 7 | 3g | 4-FC6H4 | 72 |
| 8 | 3h | 2-Naphthyl | 70 |
| aReaction conditions: under N2, 1a (0.25 mmol), 2 (0.75 mmol), Pd(OAc)2 (5 mol%), dppp (10 mol%), Na2CO3(0.75 mmol), DMF (2.0 mL), 100 ℃~120 ℃. bIsolated yield. | |||
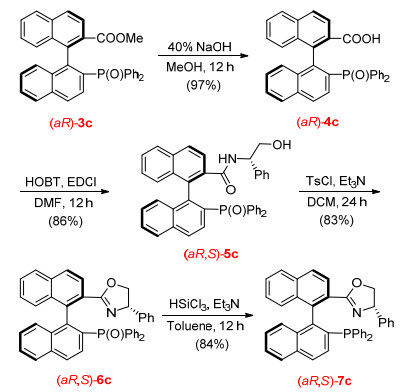
在此基础上, 我们尝试了将该方法应用于BINAP类型的N/P配体的合成中.这类配体由于配位氮原子的引入, 使得配体的电子性质以及配位方式发生了较大的变化, 从而使催化剂表现出完全不同的催化性质.该类配体在不对称氢化反应[13]、不对称氢转移反应[14]和酮的硅氢加成反应[15]中均有不凡的表现.尤其在过渡金属催化的不对称烯丙基取代反应[16]中最为常见.所以开发手性的N/P配体一直在有机化学方法学的研究中占有重要地位.因此我们按照Scheme 1所示路线进行了相应配体的合成.采用3c作为起始原料, 酯基水解后与手性氨基醇缩合得到中间体5c, 接着发生分子内关环反应, 最后经过硅氢还原得到目标产物7c.四步收率58%, 而且最终产物并未发生消旋.
本文通过对反应条件的优化, 成功实现了钯催化芳基磺酸酯与各种亚磷酸酯偶联反应, 该方法具有广泛的底物适用性.此外我们成功地将其应用于手性噁唑啉-膦配体的合成中.以芳基磺酸酯与二苯基膦氧为起始原料, 经过五步以47%的收率完成该配体的合成, 证明该方法具有较强的应用价值.
溶剂均以标准方法处理纯化.熔点在上海精密科学仪器有限公司生产的SWG X-4显微熔点仪上测定; 使用Varian Plus-400 MHz核磁共振仪进行的1H NMR, 13C NMR, 19F NMR和31P NMR谱图测试分别在400, 100, 376和165 MHz条件下获得, TMS为内标, 氘化氯仿(CDCl3)为溶剂; 使用电喷雾电离(ESI)技术记录高分辨质谱(HRMS).柱层析使用烟台化工厂生产的硅胶(300~400目).
向干燥125.0 mL的高压反应釜中加入(R)-2, 2'-二(三氟甲磺酸基)-1, 1'-联萘(20.0 mmol, 11.0 g), 10.0 mL甲醇和30.0 mL 1, 4-二氧六环.再加入醋酸钯(0.4 mmol, 89.0 mg), dppp (0.8 mmol, 330.0 mg)和N, N-二异丙基乙胺(10.3 g, 80.0 mmol).搅拌溶清后封釜, CO置气三次后, 充入2.03 MPa, 90 ℃下反应24 h.自然冷却至室温, 放气开釜, 倒出反应液并加入50.0 mL乙酸乙酯, 并将有机相水洗3次, 加入无水硫酸钠干燥, 浓缩有机相后进行柱层析分离, 得4.6 g, 收率50%.白色固体, m.p. 127~128 ℃[17]; [α]25 D-43.6 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.25 (d, J=8.4 Hz, 1H), 8.08 (t, J=8.4 Hz, 2H), 7.99 (q, J=4.4 Hz, 2H), 7.60~7.52 (m, 3H), 7.36~7.31 (m, 2H), 7.19~7.15 (m, 2H), 3.56 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 166.6, 144.3, 135.1, 133.7, 133.3, 132.5, 132.0, 130.2, 129.6, 129.3, 128.6, 128.2, 128.1, 128.0, 127.5, 127.3, 127.1, 126.8, 126.4, 126.1, 122.8, 119.6, 119.2, 116.4, 113.3 (J=318.0 Hz), 52.0; 19F NMR (376 MHz, CDCl3) δ: 2.92.
以(R)-2'-二乙氧基膦酰基-1, 1'-联萘-2-甲酸甲酯(3a)的合成为例.氮气保护下, 向Schlenk管中加入(R)-2'-三氟甲磺酸基-1, 1'-联萘-2-甲酸甲酯(1a) (115.0 mg, 0.25 mmol), 醋酸钯(2.8 mg, 0.0125 mmol), dppp (10.3 mg, 0.025 mmol), 加入2.0 mL干燥的DMF.搅拌溶解.然后加入亚磷酸二乙酯(103.0 mg, 0.75 mmol), 碳酸钠(80.0 mg, 0.75 mmol). 100 ℃下反应. 3 h后降至室温, 加入5.0 mL水猝灭反应, 乙酸乙酯萃取(5.0 mL×3), 饱和食盐水洗, 无水硫酸钠干燥, 浓缩有机相, 得到的油状物进行柱层析分离, 得106.0 mg, 收率95%.淡黄色油. [α]25 D-43.4 (c 1.00, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.24 (m, J=8.8 Hz, 1H), 8.19 (dd, J=12.4, 8.8 Hz, 1H), 8.04~8.01 (m, 2H), 7.94 (t, J=7.2 Hz, 2H), 7.54~7.49 (m, 2H), 7.26~7.20 (m, 2H), 7.10~7.03 (dd, J=20.8, 8.4 Hz, 2H), 3.83~3.73 (m, 1H), 3.69~3.57 (m, 2H), 3.48 (s, 3H), 3.45~3.39 (m, 1H), 1.02 (t, J=7.2 Hz, 3H), 0.63 (t, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.3, 142.9 (d, J=8.0 Hz), 138.8 (d, J=5.0 Hz), 134.4 (d, J=6.0 Hz), 133.3, 132.7 (d, J=16.0 Hz), 128.2~123.2 (m, Ar—C), 61.3 (d, J=6.0 Hz), 61.2 (d, J=6.0 Hz), 51.4, 15.6 (d, J=6.0 Hz), 15.1 (d, J=6.0 Hz); 31P NMR (165 MHz, CDCl3) δ: 19.2; HRMS calcd for C26H26O5P (M+ H)+ 449.1518, found 449.1517.
(R)-2'-二甲氧基膦酰基-1, 1'-联萘-2-甲酸甲酯(3b):制备方法同3a, 得100.0 mg, 收率96%.淡黄色油. [α]25 D-48.9 (c 1.02, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.22 (d, J=8.8 Hz, 1H), 8.16 (dd, J=12.0, 8.4 Hz, 1H), 8.05~8.02 (m, 2H), 7.95 (t, J=7.2 Hz, 2H), 7.56~7.50 (m, 2H), 7.28~7.23 (m, 2H), 7.07 (dd, J=44.4, 8.8 Hz, 2H), 3.50 (s, 3H), 3.36 (d, J=11.2 Hz, 3H), 3.04 (d, J=11.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.6, 143.4 (d, J=9.0 Hz), 138.7 (d, J=5.0 Hz), 134.5 (d, J=12.0 Hz), 133.3, 132.9 (d, J=16.0 Hz), 128.5~122.3 (m, Ar—C), 52.0 (d, J=6.0 Hz), 51.9 (d, J=6.0 Hz), 51.6; 31P NMR (165 MHz, CDCl3) δ: 21.6; HRMS calcd for C24H22O5P (M+H)+ 421.1205, found 449.1195.
(R)-2'-二苯基膦酰基-1, 1'-联萘-2-甲酸甲酯(3c):制备方法同3a, 得103.0 mg, 收率81%.白色固体, m.p. 102~103 ℃[12]; [α]20 D+15.3 (c 0.42, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 8.03 (d, J=8.8 Hz, 1H), 7.95~7.91 (m, 2H), 7.78 (d, J=8.8 Hz, 1H), 7.72 (d, J=8.0 Hz, 1H), 7.62 (dd, J=11.6, 8.8 Hz, 1H), 7.52~7.47 (m, 3H), 7.40~7.32 (m, 4H), 7.26~7.19 (m, 4H), 7.10~6.96 (m, 5H), 3.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.6, 144.1 (d, J=7.0 Hz), 138.1 (d, J=5.0 Hz), 134.4~125.7 (m, Ar—C), 51.7; 31P NMR (165 MHz, CDCl3) δ: 28.3.
(R)-2'-二对甲苯基膦酰基-1, 1'-联萘-2-甲酸甲酯(3d):制备方法同3a, 得103.0 mg, 收率73%.白色固体, m.p. 79~80 ℃(文献值[12] m.p. 79~80 ℃); [α]20 D+5.1 (c 0.61, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 8.02 (d, J=8.4 Hz. 1H), 7.94~7.90 (m, 2H), 7.77 (d, J=8.4 Hz, 1H), 7.71~7.63 (m, 2H), 7.52~7.48 (m, 1H), 7.41~7.33 (m, 3H), 7.23~7.16 (m, 3H), 7.11~7.07 (m, 1H), 7.03~6.95 (m, 4H), 6.85~6.83 (m, 2H), 3.52 (s, 3H), 2.31 (s, 3H), 2.21 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.7, 143.8 (d, J=7.0 Hz), 141.3, 140.9, 138.3 (d, J=5.0 Hz), 134.4~125.8 (m, Ar—C), 51.9, 21.5, 21.4; 31P NMR (165 MHz, CDCl3) δ: 28.9.
(R)-2'-二(3, 5-二甲基苯基)膦酰基-1, 1'-联萘-2-甲酸甲酯(3e):制备方法同3a, 得120.0 mg, 收率85%.白色固体, m.p. 107~108 ℃(文献值[12] m.p. 107~110 ℃); [α]20 D-8.9 (c 0.96, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 8.03 (d, J=8.8 Hz, 1H), 7.95~7.91 (m, 2H), 7.75~7.67 (m, 3H), 7.51~7.48 (m, 1H), 7.39~7.35 (m, 1H), 7.22~7.18 (m, 1H), 7.15~7.11 (m, 3H), 7.02~6.96 (m, 3H), 6.88 (d, J=12.0 Hz, 2H), 6.70 (s, 1H), 3.58 (s, 3H), 2.20 (s, 6H), 2.06 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.5, 143.7 (d, J=7.0 Hz), 138.0 (d, J=5.0 Hz), 137.4~125.6 (m, Ar—C), 51.8, 21.2, 21.1; 31P NMR (165 MHz, CDCl3) δ: 28.3.
(R)-2'-二对甲氧基苯基膦酰基-1, 1'-联萘-2-甲酸甲酯(3f):制备方法同3a, 得108.0 mg, 收率76%.白色固体, m.p. 87~88 ℃(文献值[12] m.p. 85~88 ℃); [α]20 D+14.7 (c 0.61, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 8.03 (d, J=8.8 Hz. 1H), 7.96~7.90 (m, 2H), 7.79~7.67 (m, 3H), 7.50 (t, J=8.0 Hz, 1H), 7.42~7.33 (m, 3H), 7.26~7.19 (m, 3H), 7.13~7.09 (m, 1H), 6.99 (dd, J=11.2, 8.8 Hz, 2H), 6.68 (dd, J=8.8, 2.4 Hz, 2H), 6.58 (dd, J=8.8, 2.4 Hz, 2H), 3.78 (s, 3H), 3.72 (s, 3H), 3.53 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.7, 161.5 (d, J=13.0 Hz), 143.5 (d, J=7.0 Hz), 138.4 (d, J=4.0 Hz), 134.5~123.6 (m, Ar—C), 113.4 (d, J=6.0 Hz), 113.2 (d, J=6.0 Hz), 55.2, 55.1, 51.9; 31P NMR (165 MHz, CDCl3) δ: 28.9.
(R)-2'-二对氟苯基膦酰基-1, 1'-联萘-2-甲酸甲酯(3g):制备方法同3a, 得98.0 mg, 收率72%.白色固体, m.p. 79~80 ℃(文献值[12] m.p. 79~80 ℃); [α]20 D+18.2 (c 0.78, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 8.03 (d, J=8.8 Hz. 1H), 7.99~7.92 (m, 2H), 7.77 (dd, J=20.8, 8.8 Hz, 2H), 7.64 (dd, J=11.6, 8.4 Hz, 1H), 7.53 (t, J=8.0 Hz, 1H), 7.46~7.40 (m, 3H), 7.36~7.29 (m, 2H), 7.26~7.22 (m, 1H), 7.13 (t, J=8.0 Hz, 1H), 7.00 (dd, J=33.6, 8.4 Hz, 2H), 6.87 (td, J=8.8, 2.0 Hz, 2H), 6.76 (td, J=8.4, 2.0 Hz, 2H), 3.57 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.6, 165.6 (d, J=9.0 Hz), 163.1 (d, J=12.0 Hz), 143.9 (d, J=7.0 Hz), 137.9 (d, J=4.0 Hz), 134.4~114.8 (m, Ar—C), 51.9; 19F NMR (376 MHz, CDCl3): -29.9, -30.3; 31P NMR (165 MHz, CDCl3): δ 27.2.
(R)-2'-二(2-萘基)膦酰基-1, 1'-联萘-2-甲酸甲酯(3h):制备方法同3a, 得107.0 mg, 收率70%.白色固体, m.p. 103~104 ℃; [α]20 D+46.3 (c 1.04, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 8.08 (d, J=14.0 Hz, 1H), 7.98~7.93 (m, 3H), 7.87 (d, J=8.8 Hz, 1H), 7.81~7.76 (m, 2H), 7.70~7.64 (m, 4H), 7.56~7.41 (m, 9H), 7.37 (d, J=8.0 Hz, 1H), 7.24~7.20 (m, 1H), 7.18~7.13 (m, 1H), 7.05 (d, J=8.4 Hz, 1H), 7.01 (d, J=3.6 Hz, 2H), 3.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 166.5, 144.0 (d, J=8.0 Hz), 137.8 (d, J=5.0 Hz), 134.2~125.5 (m, Ar—C), 51.8; 31P NMR (165 MHz, CDCl3) δ: 27.7; HRMS calcd for C42H30O3P (M+H)+ 613.1933, found 613.1935.
向15.0 mL甲醇和15.0 mL水的混合溶液中加入NaOH (1.2 g, 30.0 mmol), 搅拌溶解.将3c (3.0 g, 5.9 mol)加入瓶中, 体系回流12 h, 将反应液倒入20.0 mL, 2 mol/L的稀盐酸中, 用CH2Cl2萃取(20.0 mL×2).将合并的有机层用无水硫酸钠干燥, 在减压下浓缩, 得2.8 g, 收率97%[12].白色固体, m.p. 221~222 ℃; [α]20 D+164.2 (c 0.73, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 7.94~7.88 (m, 3H), 7.78~7.72 (m, 3H), 7.66~7.61 (m, 2H), 7.56~7.52 (m, 3H), 7.35~7.27 (m, 2H), 7.19~7.11 (m, 5H), 7.00~6.96 (m, 2H), 6.68~6.64 (t, J=7.2 Hz, 1H), 6.22~6.20 (d, J=8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 171.4, 142.4 (d, J=8.0 Hz), 135.5, 134.9, 133.7 (d, J=11.0 Hz), 132.9~125.9 (m, Ar—C), 123.8; 31P NMR (165 MHz, CDCl3) δ: 35.5.
将化合物4c (2.0 g, 4.0 mmol)、手性氨基醇(1.1 g, 8.0 mmol)、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(2.3 g, 12.0 mmol)和1-羟基苯并三氮唑(2.2 g, 16.0 mmol)加入到DMF (30.0 mL)中, 在室温下搅拌过夜.然后减压除去溶剂, 得到的残留物用CH2Cl2稀释, 用饱和NaCl溶液洗涤, 有机层用无水硫酸钠干燥.减压浓缩, 柱层析分离, 得到化合物5c 2.1 g, 收率86%.白色固体, m.p. 250~251 ℃(文献值[18] m.p. 254~256 ℃); [α]20 D+42.2 (c 0.80, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 9.36 (d, J=6.4 Hz, 1H), 8.03~7.97 (m, 2H), 7.85 (d, J=8.8 Hz, 1H), 7.74 (d, J=8.4 Hz, 1H), 7.68~7.63 (m, 2H), 7.58~7.42 (m, 6H), 7.26~7.09 (m, 5H), 7.00~6.95 (m, 2H), 6.85~6.76 (m, 5H), 6.49 (d, J=8.4 Hz, 1H), 6.28 (d, J=7.2 Hz, 2H), 4.95~4.91 (m, 1H), 3.86 (dd, J=12.0, 3.6 Hz, 1H), 3.67 (dd, J=12.0, 7.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 169.2, 142.9 (d, J=8.0 Hz), 139.1, 136.2, 134.9, 133.5~125.6 (m, Ar—C), 66.1, 56.6; 31P NMR (165 MHz, CDCl3) δ: 31.8.
将化合物5c (1.5 g, 2.4 mmol)和三乙胺(490.0 mg, 4.9 mmol)加入到二氯甲烷(20.0 mL)中, 降至0 ℃, 再加入对甲苯磺酰氯(930.0 mg, 4.9 mmol), 搅拌0.5 h.然后将另一份三乙胺(2.2 g, 21.9 mmol)加入到该溶液中, 回流24 h, 停止反应, 水洗, 用CH2Cl2萃取(20.0 mL 2).该有机层用无水Na2SO4干燥并减压浓缩, 柱层析分离, 得到化合物6c 1.2 g, 收率83%.白色固体, m.p. 192~193 ℃(文献值[18] m.p. 192~195 ℃); [α]20 D+9.2 (c 1.02, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.01~7.91 (m, 3H), 7.75~7.69 (m, 3H), 7.54 (t, J=6.8 Hz, 1H), 7.44~7.37 (m, 5H), 7.30~7.23 (m, 3H), 7.18~7.08 (m, 10H), 6.81~6.78 (m, 2H), 5.03 (t, J=8.4 Hz, 1H), 4.35 (dd, J=10.0, 8.4 Hz, 1H), 3.57 (t, J=8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 165.3, 143.0 (d, J=7.0 Hz), 142.1, 135.7 (d, J=5.0 Hz), 134.1~126.0 (m, Ar—C), 74.5, 69.5; 31P NMR (165 MHz, CDCl3) δ: 28.9.
氮气保护下, 将化合物6c (0.8 g, 1.3 mmol)和三乙胺(4.0 g, 40.0 mmol)加入到甲苯(20.0 mL)中, 降至0 ℃, 加入三氯硅烷(1.8 g, 13.0 mmol).此温度下搅拌0.5 h.然后回流12 h.冷却至室温后, 加入20.0 mL乙酸乙酯稀释体系, 并用饱和NaHCO3溶液猝灭.加入硅藻土, 过滤.有机层用无水硫酸钠干燥, 减压浓缩.粗产物通过柱层析纯化, 得化合物7c 653.0 mg, 收率84%.白色固体, m.p. 124~125 ℃(文献值[18] m.p. 125~127 ℃); [α]20 D-47.2 (c 0.47, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 8.17 (d, J=8.4 Hz, 1H), 8.03 (d, J=8.8 Hz. 1H), 7.93~7.89 (m, 3H), 7.52~7.41 (m, 3H), 7.31~7.09 (m, 13H), 7.07~6.99 (m, 4H), 6.68~6.66 (m, 2H), 4.89 (dd, J=10.0, 8.4 Hz, 1H), 4.12 (dd, J=10.4, 8.4 Hz, 1H), 3.43 (t, J=8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 165.6, 144.8, 144.4, 142.2, 138.4 (d, J=9.0 Hz), 138.0 (d, J=3.0 Hz), 137.8 (d, J=2.0 Hz), 135.0 (d, J=11.0 Hz), 134.2~126.2 (m, Ar—C), 74.6, 69.4; 31P NMR (165 MHz, CDCl3): δ -19.5.
辅助材料(Supporting Information) 各化合物的1H NMR、13C NMR、19F NMR、31P NMR等图谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Hostetler, K. Y. Antiviral Res. 2009, 82, A84.
(b) Thornton, P. J.; Kadri, H.; Miccoli, A.; Mehellou, Y. J. Med. Chem. 2016, 59, 10400.
(c) Wagner, C. R.; Iyer, V. V.; McIntee, E. J. Med. Res. Rev. 2000, 20, 417.
(a) Fu, W.; Tang, W. ACS Catal. 2016, 6, 4814.
(b) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029.
(a) Zhang, Y.; Yue, B.; Han, S.; Yan, L. RSC Adv. 2014, 4, 33702.
(b) Morisaki, Y.; Ouchi, Y.; Fukui, T.; Naka, K.; Chujo, Y. Tetrahedron Lett. 2005, 46, 7011.
(a) Noyori, R.; Takaya, H. Acc. Chem. Res. 1990, 23, 345.
(b) Fandrick, K. R.; Fandrick, D. R.; Reeves, J. T.; Gao, J.; Ma, S.; Li, W.; Lee, H.; Grinberg, N.; Lu, B.; Senanayake, C. H. J. Am. Chem. Soc. 2011, 133, 10332.
(c) Lu, B.; Wang, Q.; Zhao, M.; Xie, X.; Zhang, Z. J. Org. Chem. 2015, 80, 9563.
Berthod, M.; Mignani, G.; Woodward, G.; Lemaire, M. Chem. Rev. 2005, 105, 1801. doi: 10.1021/cr040652w
(a) Le Bras, J.; Muzart, J. Chem. Rev. 2011, 111, 1170.
(b) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457.
(c) Lee, C. K.; Shin, S. R.; Mun, J. Y.; Han, S.-S.; So, I.; Jeon, J.-H.; Kang, T. M.; Kim, S. I.; Whitten, P. G.; Wallace, G. G.; Spinks, G. M.; Kim, S. J. Angew. Chem., Int. Ed. 2009, 48, 5116.
(d) Shi, Y.; Peterson, S. M.; Haberaecker, W. W.; Blum, S. A. J. Am. Chem. Soc. 2008, 130, 2168.
(e) Shen, X.; Jones, G. O.; Watson, D. A.; Bhayana, B.; Buchwald, S. L. J. Am. Chem. Soc. 2010, 132, 11278.
(f) Sase, S.; Jaric, M.; Metzger, A.; Malakhov, V.; Knochel, P. J. Org. Chem. 2008, 73, 7380.
(a) Kalek, M.; Jezowska, M.; Stawinski, J. Adv. Synth. Catal. 2009, 351, 3207.
(b) Wang, T.; Sang, S.; Liu, L.; Qiao, H.; Gao, Y.; Zhao, Y. J. Org. Chem. 2014, 79, 608.
(c) Hong, G.; Mao, D.; Wu, S.; Wang, L. J. Org. Chem. 2014, 79, 10629.
(d) Deal, E. L.; Petit, C.; Montchamp, J.-L. Org. Lett. 2011, 13, 3270.
(e) Bloomfield, A. J.; Herzon, S. B. Org. Lett. 2012, 14, 4370.
(f) Zhu, J.; Mao, M.; Ji, H.-J.; Xu, J.-Y.; Wu, L. Org. Lett. 2017, 19, 1946.
(a) Gao, Y. ; Wang, G. ; Chen, L. ; Xu, P. ; Zhao, Y. ; Zhou, Y. ; Han, L. -B. J. Am. Chem. Soc. 2009, 131, 7956.
(b) Huang, C. ; Tang, X. ; Fu, H. ; Jiang, Y. ; Zhao, Y. J. Org. Chem. 2006, 71, 5020.
(c) Zhang, H. ; Li, W. ; Zhu, C. J. Org. Chem. 2017, 82, 2199.
(d) Thielges, S. ; Bisseret, P. ; Eustache, J. Org. Lett. 2005, 7, 681.
(e) Hu, G. ; Shan, C. ; Chen, W. ; Xu, P. ; Gao, Y. ; Zhao, Y. Org. Lett. 2016, 18, 6066.
(f) Xu, Q. ; Jia, X. ; Li, X. ; Sun, Q. ; Zhou, Y. ; Yin, S. ; Han, L. Chin. J. Org. Chem. 2014, 34, 1340(in Chinese).
(徐清, 贾晓娟, 李晓慧, 孙清, 周永波, 尹双凤, 韩立彪, 有机化学, 2014, 34, 1340. )
(g) Xu, Q. ; Zhao, C. ; Zhou, Y. ; Yin, S. ; Han, L. Chin. J. Org. Chem. 2012, 32, 1761(in Chinese).
(徐清, 赵长秋, 周永波, 尹双凤, 韩立彪, 有机化学, 2012, 32, 1761. )
(h) Zhang, Y. ; Wang, M. ; Cao, P. ; Liao, J. Acta Chim. Sinica 2017, 75, 794(in Chinese).
(张涌灵, 王敏, 曹鹏, 廖健, 化学学报, 2017, 75, 794. )
(a) Shen, C.; Yang, G.; Zhang, W. Org. Biomol. Chem. 2012, 10, 3500.
(b) Wu, Y.; Liu, L.; Yan, K.; Xu, P.; Gao, Y.; Zhao, Y. J. Org. Chem. 2014, 79, 8118.
(c) Zhang, X.; Liu, H.; Hu, X.; Tang, G.; Zhu, J.; Zhao, Y. Org. Lett. 2011, 13, 3478.
(d) Zhao, Y.-L.; Wu, G.-J.; Han, F.-S. Chem. Commun. 2012, 48, 5868.
(a) Kagayama, T.; Nakano, A.; Sakaguchi, S.; Ishii, Y. Org. Lett. 2006, 8, 407.
(b) Mu, X.-J.; Zou, J.-P.; Qian, Q.-F.; Zhang, W. Org. Lett. 2006, 8, 5291.
(c) Pan, X.-Q.; Zou, J.-P.; Zhang, G.-L.; Zhang, W. Chem. Commun. 2010, 46, 1721.
(d) Xu, W.; Zou, J.-P.; Zhang, W. Tetrahedron Lett. 2010, 51, 2639.
(a) Xiang, C.-B.; Bian, Y.-J.; Mao, X.-R.; Huang, Z.-Z. J. Org. Chem. 2012, 77, 7706.
(b) Hu, G.; Chen, W.; Ma, D.; Zhang, Y.; Xu, P.; Gao, Y.; Zhao, Y. J. Org. Chem. 2016, 81, 1704.
(c) Zhang, H.; Gu, Z.; Li, Z.; Pan, C.; Li, W.; Hu, H.; Zhu, C. J. Org. Chem. 2016, 81, 2122.
(d) Hu, G.; Chen, W.; Ma, D.; Zhang, Y.; Xu, P.; Gao, Y.; Zhao, Y. J. Org. Chem. 2016, 81, 2680.
(e) Zhang, B.; Daniliuc, C. G.; Studer, A. Org. Lett. 2014, 16, 250.
Zhao, Q.-Y.; Shi, M. Tetrahedron 2011, 67, 3724. doi: 10.1016/j.tet.2011.03.046
(a) Naud, F.; Malan, C.; Spindler, F.; Rüggeberg, C.; Schmidt, A. T.; Blaser, H.-U. Adv. Synth. Catal. 2006, 348, 47.
(b) Tellers, D. M.; Bio, M.; Song, Z. J.; McWilliams, J. C.; Sun, Y. Tetrahedron:Asymmetry 2006, 17, 550.
(a) Liu, D.; Xie, F.; Zhao, X.; Zhang, W. Tetrahedron 2008, 64, 3561.
(b) Nishibayashi, Y.; Takei, I.; Uemura, S.; Hidai, M. Organometallics 1999, 18, 2291.
(a) Coyne, A. G.; Guiry, P. J. Tetrahedron Lett. 2007, 48, 747.
(b) Nishibayashi, Y.; Takei, I.; Uemura, S.; Hidai, M. Organometallics 1998, 17, 3420.
(a) Fukuzumi, T.; Shibata, N.; Sugiura, M.; Yasui, H.; Nakamura, S.; Toru, T. Angew. Chem., Int. Ed. 2006, 45, 4973.
(b) Lu, Z.; Ma, S. Angew. Chem., Int. Ed. 2008, 47, 258.
(c) Nemoto, T.; Masuda, T.; Matsumoto, T.; Hamada, Y. J. Org. Chem. 2005, 70, 7172.
(d) Hou, X.-L.; Sun, N. Org. Lett. 2004, 6, 4399.
(e) Nemoto, T.; Masuda, T.; Akimoto, Y.; Fukuyama, T.; Hamada, Y. Org. Lett. 2005, 7, 4447.
Procter, D. J.; Rayner, C. M. Synth. Commun. 2000, 30, 2975. doi: 10.1080/00397910008087448
Deng, H.-P.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2009, 351, 2897. doi: 10.1002/adsc.v351:17
表 1 反应条件的优化a
Table 1. Optimization of reaction conditions
|
||||
| Entry | Solvent | Ligand | Base | Yieldb/% |
| 1 | CH3CN | dppp | Na2CO3 | 9 |
| 2 | Dioxane | dppp | Na2CO3 | Trace |
| 3 | DMSO | dppp | Na2CO3 | 47 |
| 4 | DMF | dppp | Na2CO3 | 95 |
| 5 | DMF | PPh3 | Na2CO3 | Trace |
| 6 | DMF | dppe | Na2CO3 | 11 |
| 7 | DMF | dppb | Na2CO3 | 24 |
| 8c | DMF | dppp | Na2CO3 | 95 |
| 9 | DMF | dppp | K2CO3 | Trace |
| 10 | DMF | dppp | Cs2CO3 | Trace |
| 11 | DMF | dppp | CH3COONa | 16 |
| 12 | DMF | dppp | Et3N | 15 |
| aReaction conditions: under N2, 1a (0.25 mmol), 2a (0.75 mmol), Pd(OAc)2 (5 mol%), ligand (10 mol%), base (0.75 mmol), solvent (2.0 mL), temperature is 100 ℃; reaction time is 12 h. b Isolated yield. c Reaction time is 3 h. dppp: 1, 3-bis(diphenylphosphino)propane, dppe: 1, 2-bis(diphenylphosphino)ethane, dppb: 1, 4-bis(diphenylphosphino)butane. | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 催化剂及温度的影响a
Table 2. Effects of catalyst and temperature
|
||||
| Entry | Cat. | T/℃ | t/h | Yieldb/% |
| 1 | Pd(CH3CN)2Cl2 | 100 | 4 | 92 |
| 2 | PdCl2 | 100 | 12 | 95 |
| 3 | Pd2(dba)3 | 100 | 12 | 18 |
| 4 | Pd(OAc)2 | 80 | 15 | 94 |
| 5 | Pd(OAc)2 | 50 | 12 | Trace |
| 6 | Pd(OAc)2 | 120 | 2 | 95 |
| 7c | Pd(OAc)2 | 100 | 8 | 95 |
| 8d | Pd(OAc)2 | 100 | 12 | Trace |
| aReaction conditions: under N2, 1a (0.25 mmol), 2a (0.75 mmol), Cat. (5 mol%), dppp (10 mol%), Na2CO3 (0.75 mmol), DMF (2.0 mL). bIsolated yield. cThe amount of catalyst is 1 mol%. dThe amount of catalyst is 0.5 mol%. | ||||
下载: 导出CSV
表 3 底物拓展a
Table 3. Substrate scope for the reaction
|
|||
| Entry | 3 | R | Yieldb/% |
| 1 | 3a | OEt | 95 |
| 2 | 3b | OMe | 96 |
| 3 | 3c | C6H5 | 81 |
| 4 | 3d | 4-MeC6H4 | 73 |
| 5 | 3e | 3, 5-Me2C6H3 | 85 |
| 6 | 3f | 4-MeOC6H4 | 76 |
| 7 | 3g | 4-FC6H4 | 72 |
| 8 | 3h | 2-Naphthyl | 70 |
| aReaction conditions: under N2, 1a (0.25 mmol), 2 (0.75 mmol), Pd(OAc)2 (5 mol%), dppp (10 mol%), Na2CO3(0.75 mmol), DMF (2.0 mL), 100 ℃~120 ℃. bIsolated yield. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们