 Scheme1.
Traditional synthesis of triflate esters
Scheme1.
Traditional synthesis of triflate esters
Citation:
Zheng Dongqing, Ma Haiyan, Ding Kai. A Practical Synthesis of Trifluoromethanesulfonate Esters[J]. Chinese Journal of Organic Chemistry,
2017, 37(6): 1582-1584.
doi:
10.6023/cjoc201701035

三氟甲磺酸酯的高效合成
English
A Practical Synthesis of Trifluoromethanesulfonate Esters
Abstract:
A practical synthesis of trifluoromethanesulfonate esters by reaction of orthoformate esters with triflic anhydride is described. The solvent-free method featured mild condition, short time, high yield, simple operation and broad substrate scope. The in situ generated trifluoromethanesulfonate ester is highly reactive alkylating agent, providing triflate ionic liquid in excellent yield via a one-pot procedure.
-
Key words:
- trifluoromethanesulfonate ester
- / orthoester
- / triflic anhydride
- / one-pot alkylation
-
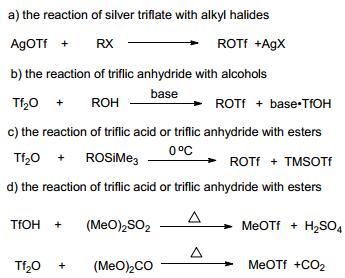
Trifluoromethanesulfonate esters (triflate esters) are versatile alkylating agent and broadly used in synthetic organic chemistry.[1] However, only a very small number of these regents are commercially available due to their instability. Therefore, many synthetic methods have been developed for the synthesis of triflate esters in the laboratory (Scheme 1).[2~6]
Scheme1.
Traditional synthesis of triflate esters
Pure triflate esters were first synthesised by the reaction of silver triflate with alkyl halide.[2] The method is clearly not economical and was soon replaced with the reaction of triflic anhydride and alcohol in the presence of acid scavengers.3 Although the method is widely used, removal of solvents and by-products from unstable products are often inefficient and laborious. Begue et al.[4] developed a simple method for the preparation of triflate esters by reaction of alkyl trimethylsilyl ethers with triflic anhydride. The low boiling by-product TMSOTf is easily removed by distillation. But the method is not economical because half of the triflic anhydride is lost. The reaction of triflic anhydride with dialkyl sulphate[5] or dialkyl carbonate[6] over-came the drawback. However, these methods required high reaction temperature and consequently suffered from a limited substrate scope. Herein, we report a practical method for the preparation of triflate esters by reaction of orthoformate esters with triflic anhydride. The solvent-free method featured mild condition (0~25 ℃), short time (15 min), high yield (up to 99%), simple operation and broad substrate scope.
1 Results and discussion
During the course of our studies on the synthesis of steroidal estrogens, [7] TfOH as a catalyst was deactivated in the presence of orthoformate esters even at room temperature, which was rationalized by the formation of triflate esters.[8] However, the attempt to prepare methyl triflate from trimethyl orthoformate was unsuccessful due to subsequent etherification with methanol. We presumed that using triflic anhydride instead of TfOH would scavenge the alcohol and suppress the undesired etherification. As expected, the reaction of triflic anhydride and methyl orthoformate provided methyl triflate in high yield at room temperature only with a trace amount of dimethyl ether (Table 1, Entry 1). Initial attempt to suppress the etherification by lowering the reaction temperature was unsuccessful (Entry 2). Fortunately, changing the addition order of reagents solved the problem because the excess Tf2O minimized the concentration of alcohol (Entry 3). The highly exothermic reaction is self-accelerating. To prevent the loss of low boiling reagents and products, the reaction was initiated at low temperature and performed at room temperature.
 Table 1.
Optimization of conditiona
Table 1.
Optimization of conditiona

Encouraged by the result, the substrate scope was investigated. Orthoformate esters were prepared from alcohols, formamide and benzoyl chloride.[9] Triflate esters were smoothly synthesized in excellent yield according to above procedure (Table 2, Entries 1~6). The only by-products are formate esters, which can be easily removed by distillation. Notably, the method can provide pure i-C3H7-OTf as a colorless liquid though the secondary triflate ester was very unstable and decomposed at room temperature after several hours (Entry 6).
Table 2.
Substrates scopea

The possible mechanism of the reaction can be described as follow (Scheme 2). In the first step, a trace amount of triflic acid in triflic anhydride reacts with the orthoformate ester to provide alcohol, formate ester and triflic ester. Subsequently, triflic anhydride plays a role of alcohol scavenger to regenerate the triflic acid.
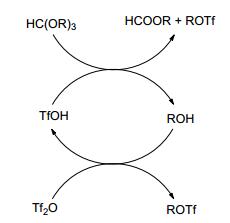 Scheme2.
Possible mechanism
Scheme2.
Possible mechanism
Triflate ester is powerful alkylating agent and, in most cases, can react with nucleophiles under mild condition, which is inert to carboxylic ester. Therefore, a one-pot, two-step protocol that bypasses the purification of the unstable and toxic alkylating agents would be possible. Triflate ionic liquid was efficiently synthesized using the protocol on 30 mmol scale (Scheme 3). The only by-product was low boiling formate ester, which was easily removed under reduced pressure to provide highly pure ionic liquid.
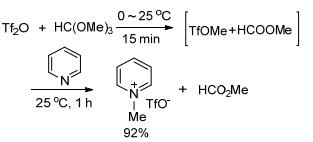 Scheme3.
One-pot synthesis of triflate ionic liquid
Scheme3.
One-pot synthesis of triflate ionic liquid
2 Conclusions
In this paper, we describe a practical synthesis of triflic esters from easily available trifilc anhydride and orthoformate esters under mild condition in excellent yield. Based on the method, a one-pot, two-step alkylation protocol was applied to synthesize triflate ionic liquid.
3 Experimental
All NMR experiments were recorded on Agilent 400 MHz NMR spectrometer or Bruker 400 MHz NMR spectrometer. Chemical shifts referenced to the residual solvent peak (1H 7.26, 13C 77.0 for CDCl3). Tf2O, trimethyl orthoformate and triethyl orthoformate were commercially available and used after distillation under reduced pressure. Other orthoformates were synthesized according to the reported method.[9]
General procedure for synthesis of triflate ester: Orthoformate ester (1.0 equiv) was added into Tf2O (1.0 equiv) at 0 ℃. The mixture was stirred at room temperature for 15 min. The pure product was obtained after a distillation under reduced pressure.
Methyl trifluorosulfonate[6]: b.p. 47 ℃/5.6 kPa; 1H NMR (400 MHz, CDCl3) δ: 4.21 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 118.7 (q, J=321.2 Hz), 61.6; 19F NMR (376 MHz, CDCl3) δ: -74.5 (s, 3F).
Ethyl trifluorosulfonate[6]: b.p. 42℃/5.6 kPa; 1H NMR (400 MHz, CDCl3) δ: 4.62 (q, J=7.1 Hz, 2H), 1.51 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 118.6 (q, J=320.8 Hz), 74.0, 15.2; 19F NMR (376 MHz, CDCl3) δ: -75.2 (s, 3F).
n-Butyl trifluorosulfonate[4]: b.p. 40℃/0.5 kPa; 1H NMR (400 MHz, CDCl3) δ: 4.54 (t, J=6.4 Hz, 2H), 1.87~1.74 (m, 2H), 1.47 (dd, J=15.0, 7.5 Hz, 2H), 0.97 (t, J=7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 118.6 (q, J=320.8 Hz), 77.5, 31.1, 18.3, 13.1; 19F NMR (376 MHz, CDCl3) δ: -75.3 (s, 3F).
n-Heptyl trifluorosulfonate[4]: b.p. 48~50℃/27 Pa; 1H NMR (400 MHz, CDCl3) δ: 4.53 (t, J=6.5 Hz, 2H), 1.90~1.76 (m, 2H), 1.44~1.28 (m, 10H), 0.89 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 118.7 (q, J=321.2 Hz), 77.8, 31.7, 29.2, 29.0, 28.8, 25.0, 22.6, 13.9; 19F NMR (376 MHz, CDCl3) δ: -75.3 (s, 3F).
i-Pentyl trifluorosulfonate[10]: b.p. 25 ℃/27 Pa; 1H NMR (400 MHz, CDCl3) δ: 4.57 (t, J=6.5 Hz, 2H), 1.82~1.75 (m, 1H), 1.72 (q, J=6.5 Hz, 2H), 0.96 (d, J=6.4 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ: 118.6 (q, J=321.2 Hz), 76.3, 37.7, 24.4, 22.0; 19F NMR (376 MHz, CDCl3) δ: -75.0 (s, 3F).
i-Propyl trifluorosulfonate[4]: b.p. 25 ℃/133 Pa; 1H NMR (400 MHz, CDCl3) δ: 5.25~5.19 (m, 1H), 1.52 (d, J=6.3 Hz, 7H); 13C NMR (101 MHz, CDCl3) δ: 118.5 (q, J=320.5 Hz), 86.4, 23.0; 19F NMR (376 MHz, CDCl3) δ: -75.9 (s, 3F).
Synthesis of N-methylpyridinium triflate: Trimethyl orthoformate (1.64 mL, 15mmol) was added into Tf2O (2.52 mL, 15mmol) at 0 ℃. After stirring for 15 min at room temperature, dry pyridine (2.73 g, 30 mmol) was slowly added into the mixture at room temperature, which was stirred for 1 h at room temperature. The moisture was heated to 60℃ for 1 h under reduced pressure to provide the pure product as a thick colorless oil (6.73 g, 92%), which solidified on standing in a refrigerator.1H NMR (400 MHz, DMSO-d6) δ: 8.97 (d, J=5.8 Hz, 2H), 8.57 (t, J=7.8 Hz, 1H), 8.12 (t, J=7.0 Hz, 2H), 4.36 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ: 145.6, 145.1, 127.8, 120.8 (q, J=323.5 Hz), 48.0; 19F NMR (376 MHz, DMSO-d6) δ: -77.8 (s, 3F).
Supporting Information 1H NMR, 13C NMR and 19F NMR spectra of all compounds. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
-
-
[1]
(a) Ulibarri, G. ; Choret, N. ; Bigg, D. C. H. Synthesis 1996, 1286.
(b) Wang, S. F. ; Zhang, A. J. Org. Prep. Proced. Int. 2008, 40 (3), 293.
(c) Chen, Z. J. ; Xi, H. W. ; Lim, K. H. ; Lee, J. M. Angew. Chem. , Int. Ed. 2013, 52(50), 13392.
(d) Dang, H. ; Mailig, M. ; Lalic, G. Angew. Chem. , Int. Ed. 2014, 53(25), 6473.
(e) Zhao, P. ; Yan, X. Y. ; Yin, H. ; Xi, C. J. Org. Lett. 2014, 16(4), 1120. -
[2]
(a) Gramstad, T.; Haszeldine, R. N. J. Chem. Soc. 1956, 173. 1.
(b) Chapman, R. D.; Andreshak, J. L.; Herrlinger, S. P.; Shackelford, S. A.; Hildreth, R. A.; Smith, J. P. J. Org. Chem. 1986, 51(20), 3792. -
[3]
Baraznenok, I. L.; Nenajdenko, V. G.; Balenkova, E. S. Tetrahedron 2000, 56(20), 3077. doi: 10.1016/S0040-4020(00)00093-4
-
[4]
Aubert, C.; Begue, J. P. Synthesis 1985, (8), 759.
-
[5]
Beard, C. D.; Baum, K.; Grakausk, V. J. Org. Chem. 1973, 38(21), 3673. doi: 10.1021/jo00961a003
-
[6]
Ignat'ev, N. V.; Barthen, P.; Kucheryna, A.; Willner, H.; Sartori, P. Molecules 2012, 17(5), 5319.
-
[7]
Zheng, D.-Q.; Jing, Y.; Zheng, B.-Y.; Ye, Y.-F.; Xu, S.; Tian, W.-S.; Ma, H.-Y.; Ding, K. Tetrahedron 2016, 72(17), 2164. doi: 10.1016/j.tet.2016.03.002
-
[8]
(a) Padmapriya, A. A.; Just, G.; Lewis, N. G. Synth. Commun. 1985, 15(12), 1057.
(b) Trujillo, J. I.; Gopalan, A. S. Tetrahedron Lett. 1993, 34(46), 7355.
(c) Yoshino, T.; Togo, H. Synlett 2005. -
[9]
Ohme, R.; Schmitz, E. Justus Liebigs Ann. Chem. 1968, 716, 207. doi: 10.1002/(ISSN)1099-0690
-
[10]
Anderson, G. L.; Harruna, I. Synth. Commun. 1987, 17(1), 111. doi: 10.1080/00397918708063910
-
[1]
-


 下载:
下载:


 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 24
- 文章访问数: 1642
- HTML全文浏览量: 355
 下载:
下载:
