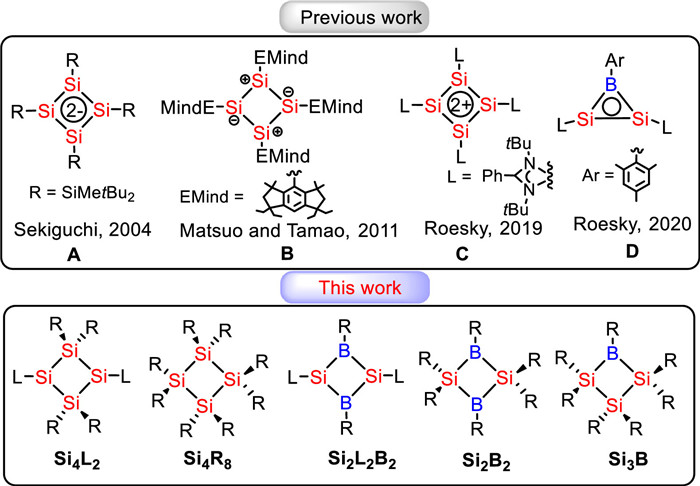
Scheme 1.
Some previously reported Si4/Si2B complexes and the proposed frameworks in this work.

Aromaticity, a significant concept in chemistry, has attracted substantial interests from both experimentalists and theoreticians due to its numerous manifestations [1-11]. In general, the terms π-aromaticity and σ-aromaticity are used to describe unsaturated and saturated systems, respectively [12,13]. Therefore, it becomes an interesting question whether it is possible to locate unsaturated σ-aromatics or saturated π-aromatics? Indeed, previous studies have shown that σ-aromaticity can dominate in unsaturated systems [14]. In the past eight years, our group has focused on studying σ-aromaticity in various unsaturated systems including three-membered rings (3MRs) of cyclopropametallapentalenes from group 7 to 9 [15,16] as well as fully unsaturated cyclopropene systems [17]. Additionally, we have also investigated σ-aromaticity in Se-containing complexes [18]. In 2020, it was reported that unsaturated cyclopropametallapentalenes exhibit both σ-aromaticity in singlet and triplet states via DFT calculations, which is referred to as "adaptive σ-aromaticity" [19]. In contrast, it is rarely reported that π-aromaticity could be dominating in saturated systems [20].
On the other hand, cyclic organosilanes [21-30], especially analogues of cyclobutadiene (CBD, including 2π-aromatic CBD2+ and 6π-aromatic CBD2−), have attracted considerable attention due to their unique electronic properties and reactivity [31,32]. Sekiguchi and co-workers reported the silicon analogue A (Scheme 1) of the CBD2− with 6π electrons, which exhibited non-aromatic characteristics due to the strong ring distortion and significantly different Si-Si distances [33]. A neutral silicon analogue of CBD, B (Scheme 1), reported by Matsuo and Tamao in 2012, possesses non-aromaticity owing to the lack of electron delocalization on the ring caused by the charge separation [34]. After that, a charge-localized nonaromatic tetrasilacyclobutadiene dication was isolated by Inoue and Driess [35]. In 2013, an aromatic planar and rhombic four-membered cycle was synthesized by So and co-workers, exhibiting a unique delocalization of n, π, and σ electrons [36]. In 2019, the first aromatic silicon analogue of the cyclobutadiene dication, C (Scheme 1), was synthesized and characterized by Roesky and co-workers [37], which has been over 50 years since the synthesis of aromatic carbon-based CBD-dications [38,39]. In addition, several silicon analogues of 1,3-cyclobutanedilyls have been isolated, including tetrasilabicyclo[1.1.0]butanes with a 1,3-Si–Si σ bond [40-43] or a 1,3-Si–Si π bond [44-46]. Recently, Roesky, Jemmis, and Stalke co-reported a neutral 2π-aromatic three-membered disilaborirane (Si-B-Si ring, D in Scheme 1) and succeeded in further converting it into a four-membered heterocycle 1-aza-2,3-disila-4-boretidine (BSi2N-Ring) [47]. Theoretically, as early as 1996, Clark and co-workers reported that fragments with low pseudo-π* levels in C2R2PF2+ and C2R2SiF2 could enhance their π conjugation, resulting in a σ*-aromaticity that is more pronounced than their saturated analogs [48,49]. Thereafter, Jemmis and co-workers reported in 2013 that a hypothetical model, P3F92−, could exhibit pseudo-π* 2π-aromaticity, where the aromaticity arises solely from the overlap of pseudo-π* orbitals [50]. However, further analysis indicated that π-aromaticity is not the major contributor in the 3MR [20]. A counterpart can be traced back to the deltate ion, C3O32−, reported by West and his colleagues in 1979 [51]. Furthermore, the origin of the aromaticity in the well-known cyclopropenyl cation, C3H3+, is also similar to the two aforementioned examples. Although experimental and theoretical chemists have reported many aromatic Si4 rings, the discovery of a completely saturated species with dominating π-aromaticity is particularly rare [20]. Our persistent curiosity about aromaticity [52] has driven us to explore the π-aromaticity in saturated rings. Herein, we demonstrate that π-aromaticity could be dominating in several fully saturated four membered rings (4MRs), including Si4-rings, Si2B2-rings, and Si3B-rings (Scheme 1).
Inspired by the recent work of Cui [20], we have designed several model molecules as shown in Fig. 1, incorporating the strategy of introducing fluorine substituents to enhance the overlap between the pseudo-π* orbitals of the atoms participating in aromaticity.
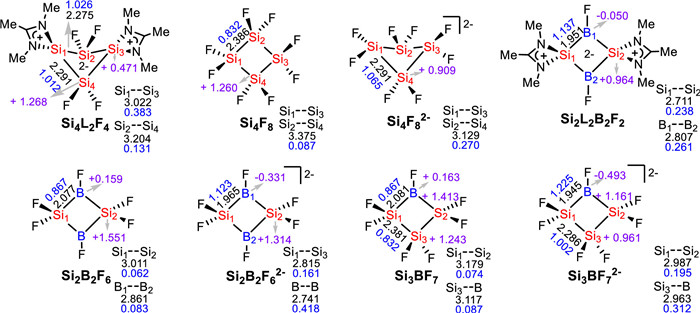
By the density functional theory (DFT) calculations, the optimized these saturated 4MRs (Si4L2F4, Si4F8, Si4F82−, Si2L2B2F2, Si2B2F6, Si2B2F62−, Si3BF7, and Si3BF72−) in the singlet ground states are shown in Fig. 1. The results suggest that Si4L2F4, Si4F8, and Si4F82− exhibit similar Si-Si bonding (bond length and Wiberg bond index (WBI) [53]) to C, a previously reported aromatic CBD2+ anolog [37]. Specifically, the distance of Si-Si bond in Si4-rings are between 2.286 Å and 2.386 Å, which is almost intermediate between Si-Si single (ca. 2.35 Å) [54] and double bonds (2.12–2.25 Å) [55], [56]. WBI values of Si-Si bond are between 0.832 and 1.065. Furthermore, the distance of Si-B bond in Si2B2-/Si3B-rings are between 1.945 Å and 2.381 Å, and the corresponding WBI values are between 1.225 and 0.832. In comparison, the WBIs between the transannular two atoms (Si-Si/Si-B/B-B) are significantly weaker than their corresponding σ-bonding (Si-Si: 0.832; Si-B: 0.867). The analysis of atoms in molecules (AIM) also supports it in Fig. S1 (Supporting information). In addition, different ligands and substituents on Si atoms in saturated 4MRs result in different planarity, in which Si4F82− has the largest degree of puckering (∠Si1-Si2-Si3-Si4: 29.6°). Except for Si4F82− (29.0°) and Si3BF72− (7.1°), the 4MRs of all other compounds are completely planar. Calculations suggest that the relative electronic energies of these saturated 4MRs in the lowest singlet states are thermodynamically more stable than those in the lowest triplet states (ΔEST, Table S1 in Supporting information), excluding a diradical ground state. Frontier molecular orbital analysis (Fig. S2 in Supporting information) shows that HOMOs of Si4L2F4, Si4F82−, Si2L2B2F2, Si2B2F62−, and Si3BF72− are the π orbitals of the 4MRs, enabling them as aromatic candidates. Meanwhile, NAO calculations are employed to provide further composition analysis of the HOMOs. Specifically, all HOMOs of them mainly correspond to the p orbitals of these Si/B atoms in 4MRs (Fig. S3 in Supporting information, Si4L2F4: 79.4%; Si4F82−: 86%; Si2L2B2F2: 81.8%; Si4F82−: 87.6%; Si3BF72−: 86.1%) with some negligible contributions of other atoms. Moreover, their HOMO-LUMO gaps are between 2.2 eV and 3.8 eV.
In addition, the bond length alternation (ΔBL) is calculated by comparing the longest Si-Si/Si-B bond length and the shortest one, providing a simple measure to evaluate the extent of bond delocalization. The calculation results (Table S1) indicate that the Si-Si and Si-B in the 4MRs of these models (Si4L2F4: 0.016 Å, Si4F82−, Si2L2B2F2, Si2B2F62−: 0.0 Å) are highly equalized, which suggests delocalized Si-Si/Si-B bonds.
In general, the nucleus-independent chemical shifts (NICS), a widely accepted index in magnetic perspective, was used for assessing aromaticity and the distinctively (positive) negative value indicates (anti)aromaticity. Here, in order to compare the aromaticity of 4MRs in these saturated models, NICS(0) and NICS(1) are employed. As shown in Fig. S4 (Supporting information), the NICS(0) and NICS(1) values for Si4 system are −19.2, −12.6, and −22.8, −13.4 ppm, respectively, indicating their aromatic character. The neutral Si2L2B2F2 model compound has NICS(0) and NICS(1) values of −16.7 and −11.2 ppm, respectively. The NICS(0) and NICS(1) values for the dianionic Si2B2/Si3B compounds are similar to those of the Si4 ring, suggesting they are aromatic too. In comparison, NICS analysis was also conducted on the pure organic saturated four-membered carbon ring C4F82−, and the results also indicated its aromatic character. Note that the previously reported 2π-aromatic C has slightly less negative NICS values (−14.8, −10.5 ppm) in comparison with other Si-containing rings. In addition, the NICS-grid plots of Si4F8, Si2B2F6, and Si3BF7 show that they are nonaromatic, antiaromatic, and antiaromatic, respectively (Fig. S5 in Supporting information).
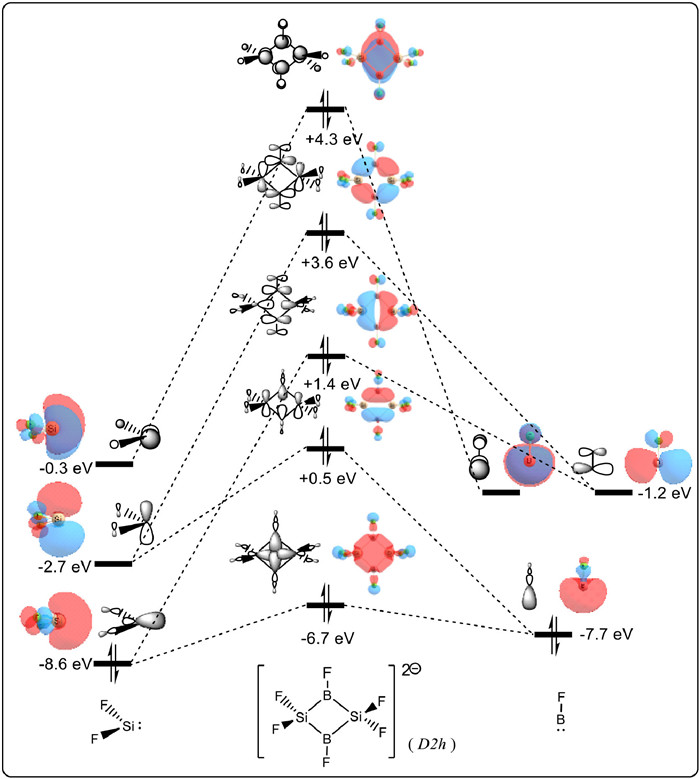
A fascinating phenomenon is observed when comparing Si2B2F6 and Si2B2F62−. This phenomenon involves a remarkable reversal in aromaticity, namely from antiaromaticity to aromaticity. Why can a saturated σ-framework achieve π-aromaticity? How does the corresponding molecular orbital change? Thus, to examine the individual orbital contributions of Si2B2F6 and Si2B2F62− to the aromaticity, CMO (canonical molecular orbital)-NICS calculations were performed. As shown in Fig. 2 and Fig. S6 (Supporting information), the NICS(0) value of the HOMO in the Si2B2F6 is +22.4 ppm, which accounts for 91.1% of its total NICS(0). Additionally, the NICS(0) of the dianion's HOMO is −22.7 ppm, making it the dominating contributor to the total NICS(0). This indicates a reversal from σ-antiaromatic to π-aromatic behavior. Moreover, combined with the analysis of the frontier molecular orbitals (Fig. S2 in Supporting information), it can be inferred that the π-character of the LUMO gains two additional electrons and become the new HOMO, which is the origin for the achievement of π-aromaticity in Si2B2F62−.

To further clarify the unique aromatic system, namely the π-aromaticity dominant in saturated scaffolding, the analysis of molecular orbital (MO) interactions is employed. In Fig. 3 and Fig. S7 (Supporting information), the fragmentation of Si2B2F6 and Si2B2F62− into [:SiF2] fragments and [:BF] fragments clearly reveals the formation of π orbitals in the σ-skeletons through MO interactions. In previous studies, the aromaticity triggered solely by a delocalizing p orbital (HOMO) was described by pseudo-π* aromaticity. The pseudo-aromatic phosphirenes (C2R2PL3) and (PF3)32− are examples reported in the literature [57]. The former involves the participation of pseudo-π* orbitals from fragment PL2L (L2 = catecholate, L = -Ph or -CN) and p orbitals from fragment CH, while the latter involves the overlap of pseudo-π* orbitals from three PF3 fragment molecules. Similarly, the HOMO of the model molecule Si2B2F62− is derived from the collective contribution of pseudo-π* orbitals of fragments [:SiF2] and [:BF], serving as another example of pseudo-π* aromaticity. Furthermore, the subsequent EDDBπ/σ separation analysis in the following text also provides the support for such π-aromaticity dominant in a σ-framework. Although the HOMO of the parent Si2B2F6 molecule is a delocalized σ orbital, the LUMO is formed by the interaction between π*-typed fragment orbitals. This provides the possibility of reversing the aromaticity of a saturated 4MRs by filling the LUMO with two additional electrons, which is in complete agreement with the CMO—NICS analysis. Additionally, as shown in Fig. S8 (Supporting information), the neutral Si2L2B2F2 molecule containing the same Si2B2 ring is also subjected to the same analysis of fragment orbital interactions. The results showed that the interaction between the π* orbitals of [:Si(NMe)2C(Me)]+ and [:BF] is the origin for the pseudo-π* 2π-aromaticity formation. Meanwhile, it is worth noting that such a strategy is applicable in saturated Si4-rings (Si4F82−, Fig. S9 in Supporting information) as well.
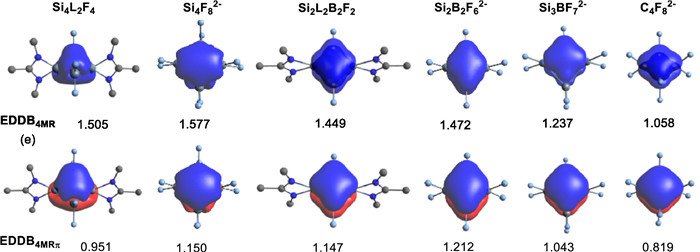
In addition, the electron density of delocalized bonds (EDDB) is used to further quantify the aromaticity of the 4MR in Si4L2F4, Si4F82−, Si2L2B2F2, Si2B2F62−, and Si3BF72−. The larger values of EDDB indicate a higher degree of electron delocalization and stronger aromaticity. For comparison, the EDDB values of the model C4F82− is also calculated. As shown in Table 1, the calculation results show that Si4 compounds (Si4L2F4, Si4F82−) have more delocalized electrons on the 4MR (1.505–1.577e), while the number of delocalized electrons on the Si2B2-/Si3B-rings (1.237–1.472e) decreases as the number of boron atoms decreases. In addition, compared to the 4MR compounds containing heavier atom Si, C4F82− has less delocalized electrons, which can be attributed to the stronger electron accommodation ability of the heavier atoms. However, it is worth noting that the mentioned effects for Si2B2-/Si3B-rings are relatively small here due to the involvement of empty p orbitals in boron atoms. As we all know, aromaticity can be classified into different types based on the electronic nature of the main contributing electrons. Therefore, the aromaticity can be further classified by separating the σ and π electrons in the delocalized system. Further EDDBπ/σ separation analysis shows that the π-aromaticity of these 4MRs (63.2%–84.3%) is dominated, which is supported by the visual isosurface plots in Fig. 4. Note that Si4L2F4 has a relatively small EDDB4MR-π value (0.951e, 63.2%), which may be related to the puckered configuration of it. Additionally, the EDDB analysis results of Si4F8, Si2B2F6, Si3BF7, and C4F8 are shown in Table S2 (Supporting information).
MCI (multicenter indices) can also provide an insight into the extent of electron sharing among atoms within the ring. Generally, rings with higher positive MCI values are more likely to exhibit stronger aromaticity. Herein, it is applied to the aromaticity analyses of these systems containing heavy atoms. In Table 1, the calculated results show that the MCI values of these saturated 4MRs (0.384–0.482) comparable to those of Si4 rings in aromatic compound C (0.449) reported by Roesky, further supporting 2π-aromaticity of these compounds.
To further understand the bonding of the 2π-aromatic Si2B2F62−, the adaptive natural density partitioning (AdNDP) analysis was performed. Not surprisingly, the result shows that the total 58 electrons can be partitioned into six s-type and twelve p-type lone pairs (LPs) on the F atoms, two B-F σ bonds, four Si-F σ bonds, four Si-B σ bonds, and one delocalized 4c-2e π bond (Fig. S10 in Supporting information). This analysis indicates that Si2B2F62− is a 2π-aromatic compound, which also provides another support for π-aromaticity in the σ-framework.
To examine the substituent effect on such pseudo-π* 2π-aromaticity, we perform additional calculations for common 4MR skeleton compounds containing amidinate ligands experimentally (Si2L2B2F2, L = MeC(MeN)2). As shown in Table S3 (Supporting information), when the substitute F in boron atom is replaced by H, Me, Ph, SiH3, CF3, NH2, and PMe2, the aromaticity of 4MRs does not change significantly, which is supported by EDDB4MR values (between 1.257e and 1.452e). The π-aromaticity of these Si2B2-rings (EDDB4MR-π/EDDB4MR: 75.5%−84.9%) is dominated too.
In summary, we have demonstrated here that π-aromaticity in several fully saturated 4MRs, which is traditionally dominated by σ-aromaticity. Specifically, the 4MRs in model complexes Si4L2F4, Si4F82−, Si2L2B2F2, Si2B2F62−, and Si3BF72− are all 2π-aromatic in the singlet ground states via DFT calculations. As the LUMO of the neutral Si2B2F6 is π-type, the corresponding dianion Si2B2F62− becomes π-aromatic due to the introduction of two electrons into such a π-type LUMO. Thus, the achievement of π-aromaticity in those saturated dianionic Si4-/Si2B2-/Si3B-rings could be attributed to the π-type LUMO of the parent neutral counterparts. Our findings provide a novel pathway for achieving π-aromaticity in fully saturated systems. Together with the previous findings on σ-aromaticity in fully unsaturated systems [14,15], such crossing aromaticity is complete.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We acknowledge financial support from the Chinese National Natural Science Foundation (No. 22231009) and the University Development Fund at the Chinese University of Hong Kong, Shenzhen (No. UDF01003116).
Supplementary material associated with this article can be found, in the online version, at doi:
P.J. Garratt, Aromaticity, Wiley, Hoboken, 1986.
V.I. Minkin, M.N. Glukhovtsev, B.Y. Simkin, Aromaticity and Antiaromaticity: Electronic and Structural Aspects, Wiley, Hoboken, 1994.
P.v.R. Schleyer, Chem. Rev. 101 (2001) 1115–1118. doi: 10.1021/cr0103221
X.Y. Cao, Q. Zhao, Z. Lin, H. Xia, Acc. Chem. Res. 47 (2014) 341–354. doi: 10.1021/ar400087x
C. Zhu, H. Xia, Acc. Chem. Res. 51 (2018) 1691–1700. doi: 10.1021/acs.accounts.8b00175
M. Rosenberg, C. Dahlstrand, K. Kilså, H. Ottosson, Chem. Rev. 114 (2014) 5379–5425. doi: 10.1021/cr300471v
J. Oh, Y.M. Sung, Y. Hong, D. Kim, Acc. Chem. Res. 51 (2018) 1349–1358. doi: 10.1021/acs.accounts.7b00629
D. Chen, Y. Hu, H. Xia, Chem. Rev. 120 (2020) 12994–13086. doi: 10.1021/acs.chemrev.0c00392
Y. Li, H. Chen, L. Qu, R. Bai, Y. Lan, Chin. Chem. Lett. 30 (2019) 2249–2253. doi: 10.3390/en12122249
C. Liu, B. Qiao, L. Qu, et al., Org. Chem. Front. 9 (2022) 4009–4015. doi: 10.1039/d2qo00797e
Y. Huang, D. Chen, J. Zhu, Z. Sun, Chin. Chem. Lett. 33 (2022) 2139–2142. doi: 10.1016/j.cclet.2021.08.038
F.D. Proft, P. Geerlings, Chem. Rev. 101 (2001) 1451–1464. doi: 10.1021/cr9903205
M. Solà, Nat. Chem. 14 (2022) 585–590. doi: 10.1038/s41557-022-00961-w
D. Chen, Q. Xie, J. Acc. Chem. Res. 52 (2019) 1449–1460. doi: 10.1021/acs.accounts.9b00092
C. Zhu, X. Zhou, H. Xing, et al., Angew. Chem. Int. Ed. 54 (2015) 3102–3106. doi: 10.1002/anie.201411220
Y. Hao, J. Wu, J. Zhu, Chem. Eur. J. 21 (2015) 18805–18810. doi: 10.1002/chem.201502390
J. Wu, X. Liu, Y. Hao, et al., Chem. Asian J. 13 (2018) 3691–3696. doi: 10.1002/asia.201801279
X. Zhou, J. Wu, Y. Hao, et al., Chem. Eur. J. 24 (2018) 2389–2395. doi: 10.1002/chem.201703870
Y. Huang, C. Dai, J. Zhu, Chem. Asian J. 15 (2020) 3444–3450. doi: 10.1002/asia.202000900
Y. Li, S. Dong, J. Guo, et al., J. Am. Chem. Soc. 145 (2023) 21159–21164. doi: 10.1021/jacs.3c06555
V.Y. Lee, A. Sekiguchi, M. Ichinohe, N. Fukaya, J. Organomet. Chem. 611 (2000) 228–235. doi: 10.1016/S0022-328X(00)00438-1
Y.F. Yang, G.J. Cheng, J. Zhu, et al., Chem. Eur. J. 18 (2012) 7516–7524. doi: 10.1002/chem.201103443
C.B. Yildiz, K.I. Leszczyńska, S. González-Gallardo, et al., Angew. Chem. Int. Ed. 59 (2020) 15087–15092. doi: 10.1002/anie.202006283
K. Abersfelder, A.J.P. White, H.S. Rzepa, D. Scheschkewitz, Science 327 (2010) 564–566. doi: 10.1126/science.1181771
A. Tsurusaki, C. Iizuka, K. Otsuka, S. Kyushin, J. Am. Chem. Soc. 135 (2013) 16340–16343. doi: 10.1021/ja409074m
J. Keuter, K. Schwedtmann, A. Hepp, et al., Angew. Chem. Int. Ed. 56 (2017) 13866–13871. doi: 10.1002/anie.201705787
T. Iwamoto, T. Abe, K. Sugimoto, et al., Angew. Chem. Int. Ed. 58 (2019) 4371–4375. doi: 10.1002/anie.201900824
J. Keuter, A. Hepp, C.G. Daniliuc, M. Feldt, F. Lips, Angew. Chem. Int. Ed. 60 (2021) 21761–21766. doi: 10.1002/anie.202104341
K. Schwedtmann, M. Quest, B.J. Guddorf, et al., Chem. Eur. J. 27 (2021) 17361–17368. doi: 10.1002/chem.202103101
T. Iwamoto, N. Akasaka, S. Ishida, Nat. Commun. 5 (2014) 5353. doi: 10.1038/ncomms6353
S. Kyushin, Organosilicon clusters. In: V.Y. Lee (Ed.), Organosilicon Compounds: Theory and Experiment (Synthesis), Academic Press, New York, 2017, p. 69.
Y. Heider, D. Scheschkewitz, Chem. Rev. 121 (2021) 9674–9718. doi: 10.1021/acs.chemrev.1c00052
V.Y. Lee, K. Takanashi, T. Matsuno, M. Ichinohe, A. Sekiguchi, J. Am. Chem. Soc. 126 (2004) 4758–4759. doi: 10.1021/ja030662z
K. Suzuki, T. Matsuo, D. Hashizume, et al., Science 331 (2011) 1306–1309. doi: 10.1126/science.1199906
S. Inoue, J.D. Epping, E. Irran, M. Driess, J. Am. Chem. Soc. 133 (2011) 8514–8517. doi: 10.1021/ja2033475
S.H. Zhang, H.W. Xi, K.H. Lim, C.W. So, Angew. Chem. Int. Ed. 52 (2013) 12364–12367. doi: 10.1002/anie.201305567
X. Sun, T. Simler, R. Yadav, R. Köppe, P.W. Roesky, J. Am. Chem. Soc. 141 (2019) 14987–14990. doi: 10.1021/jacs.9b08018
H.H. Freedman, A.M. Frantz, J. Am. Chem. Soc. 84 (1962) 4165–4167. doi: 10.1021/ja00880a049
G.A. Olah, J.M. Bollinger, A.M. White, J. Am. Chem. Soc. 91 (1969) 3667–3669. doi: 10.1021/ja01041a052
S. Masamune, Y. Kabe, S. Collins, D.J. Williams, R. Jones, J. Am. Chem. Soc. 107 (1985) 5552–5553. doi: 10.1021/ja00305a053
R. Jones, D.J. Williams, Y. Kabe, S. Masamune, Angew. Chem. Int. Ed. 25 (1986) 173–174. doi: 10.1002/anie.198601731
K. Takanashi, V.Y. Lee, M. Ichinohe, A. Sekiguchi, Chem. Lett. 36 (2007) 1158–1159. doi: 10.1246/cl.2007.1158
T. Nukazawa, T. Iwamoto, J. Am. Chem. Soc. 142 (2020) 9920–9924. doi: 10.1021/jacs.0c03874
T. Nukazawa, T. Iwamoto, Chem. Commun. 57 (2021) 9692–9695. doi: 10.1039/d1cc04332c
T. Koike, R. Osawa, S. Ishida, T. Iwamoto, Angew. Chem. Int. Ed. 61 (2022) e2021175.
S. Kyushin, Y. Kurosaki, K. Otsuka, et al., Nat. Commun. 11 (2020) 4009. doi: 10.1038/s41467-020-17815-z
S.K. Sarkar, R. Chaliha, M.M. Siddiqui, et al., Angew. Chem. Int. Ed. 59 (2020) 23015–23019. doi: 10.1002/anie.202009638
A. Göller, H. Heydt, T. Clark, J. Org. Chem. 61 (1996) 5840–5846. doi: 10.1021/jo960387h
A. Göller, T. Clark, J. Mol. Model. 6 (2000) 133–149. doi: 10.1007/PL00010724
C.P. Priyakumari, E.D. Jemmis, J. Am. Chem. Soc. 135 (2013) 16026–16029. doi: 10.1021/ja408308g
R. West, D. Eggerding, J. Perkins, D. Handy, E.C. Tuazon, J. Am. Chem. Soc. 101 (1979) 1710–1714. doi: 10.1021/ja00501a010
Q. Zhu, S. Chen, D. Chen, et al., Fundam. Res. 3 (2023) 926–938. doi: 10.1016/j.fmre.2023.04.004
K.B. Wiberg, Tetrahedron 24 (1968) 1083–1096. doi: 10.1016/0040-4020(68)88057-3
H.B. Yokelson, A.J. Millevolte, G.R. Gillette, R. West, J. Am. Chem. Soc. 109 (1987) 6865–6866. doi: 10.1021/ja00256a058
P.P. Power, Chem. Rev. 99 (1999) 3463–3504.
T. Iwamoto, S. Ishida, Multiple bonds with silicon: recent advances in synthesis, structure, and functions of stable disilenes. In: D. Scheschkewitz, (Ed.), Functional Molecular Silicon Compounds Ⅱ. Structure and Bonding, Springer International Publishing: Cham, 2014; pp 125–202.
S. Sase, N. Kano, T. Kawashima, J. Am. Chem. Soc. 124 (2002) 9706–9707. doi: 10.1021/ja026107z

Scheme 1 Some previously reported Si4/Si2B complexes and the proposed frameworks in this work.

Figure 1 Structural parameters of 4MR compounds at B3LYP-D3/def2-SVP level. Bond lengths (black) are given in Å; Wiberg bond index (blue). The natural population analysis of the charges (purple).

Figure 2 Key occupied MOs and their contributions to NICS(0) (ppm) in model complexes Si2B2F6 and Si2B2F62−.

Figure 3 Fragment MOs interaction diagram for model molecule Si2B2F62− (isovalue = 0.05 a.u.).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
