图 1.
获得手性环丙烷的对映选择性策略
Figure 1.
Enantioselective protocols to obtain enantiopure cyclopropanes

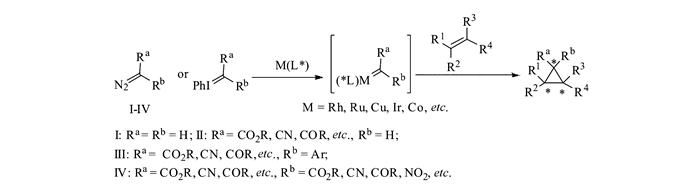
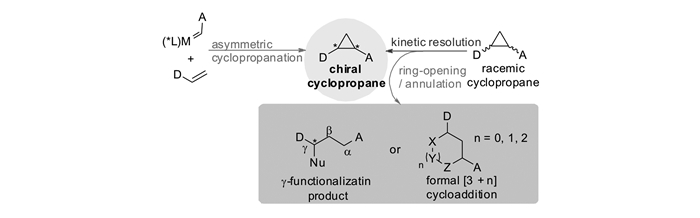
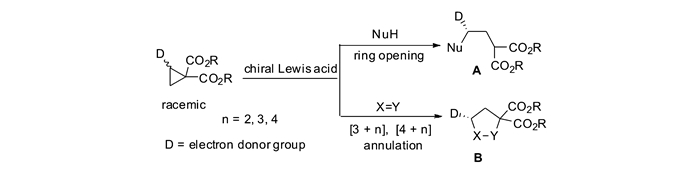
含有推-拉电子(DA)体系的环丙烷化合物是一类在有机合成中非常有用的合成砌块,长期以来一直受到化学家们的关注。人们发展了许多方法来构建这类环丙烷,特别是手性环丙烷,并使其与各种各样的亲核试剂发生开环反应,从而合成结构丰富多样的重要化合物分子[1-7]。获得高对映选择性的手性环丙烷主要有2种途径:1)直接通过不对称环丙烷化反应合成;2)针对环丙烷外消旋体的动力学拆分。在手性Lewis酸催化剂的作用下,当亲核试剂对外消旋的DA环丙烷发生动力学拆分时,可以在获得手性环丙烷的同时,生成γ-官能团化的不对称开环手性化合物;当DA环丙烷与不饱和体系发生形式[3+n]反应时,还可以获得手性的环状化合物(图 1)。
实现不对称环丙烷化反应的途径有很多,其中利用金属卡宾与烯烃发生不对称环丙烷化反应是最直接、最有效的策略之一[8-10]。由于卡宾物种的高度活泼性,人们往往采用重氮化合物、芳基碘叶立德等作为卡宾前驱体化合物,与过渡金属化合物原位生成金属卡宾物种,再参与反应。常用的金属有铑、钌、铜、铱、钴等。重氮化合物上取代基的电子特性对其反应活性有较大的影响。当重氮化合物上无拉电子稳定基团时,由于其反应活性相当高,保存也比较困难,针对其研究较少[11];当重氮化合物上连有单拉电子取代基(Ⅱ)或者推-拉电子取代基(Ⅲ)时,其在室温条件下有一定的稳定性,方便制备和保存,同时在过渡金属催化下易于生成金属卡宾活性物种,因此被广泛地应用于DA环丙烷的合成研究中;当重氮化合物含有两个拉电子基团(Ⅳ)时,化合物变得十分稳定,往往需要在剧烈条件下才能分解,从而使得其不对称反应比较困难。人们发现,当使用芳基碘叶立德作为该类重氮化合物的替代物,可以在较温和的条件下实现金属卡宾物种的生成。这类含有双拉电子基团的金属卡宾物种,例如丙二酸酯衍生物,与烯烃反应能够生成双酯基DA环丙烷。这类环丙烷由于其带有两个酯基活化基团,易于与金属Lewis酸螯合,使得张力环碳碳键更易被活化,容易发生多种形式的转化(图 2)。
对于双酯基DA环丙烷,其发生动力学拆分或者动态动力学拆分,可以作为灵活多变的有机合成子。通过与亲核试剂的不对称开环和[3+n]环化反应,这些反应为获得具有光学活性的γ-官能团化的开环结构A和环状分子B提供了简洁高效的合成路线。这种开环和环化策略被作为关键步骤,成功地应用于许多天然产物和药物分子的全合成中(图 3)[12-14]。
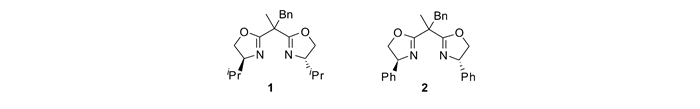
1966年,Nazaki小组[15-16]报道了用手性水杨醛亚胺与铜配合物催化重氮乙酸乙酯与苯乙烯的不对称环丙烷化反应。随后,针对烯烃与金属卡宾的不对称环丙烷化反应取得了长足的进展。Pfaltz等[17-19]报道了铜/半咕啉催化的烯烃环丙烷化反应并提出了该反应的手性诱导模型;Doyle小组[20]在铑催化体系下实现了大位阻酯基铑卡宾与末端烯烃的不对称环丙烷化反应;Evans等[21]利用手性双噁唑啉/铜催化体系实现了烯烃与重氮乙酸酯的不对称环丙烷化反应,取得了99%ee(对映体过量);Katsuki小组[22]利用手性联吡啶/铜催化剂实现了反式烯烃的不对称环丙烷化,虽然对映选择性优秀,但是非对映选择性不高;随后,他们[23]又发展了Ir-Salen催化剂,实现了顺式烯烃的不对称环丙烷化,取得了专一的非对映选择性和89%ee;Hayashi小组[24]发展了手性双烯配体/铑催化的重氮丙二酸酯和末端烯烃的不对称反应,合成了手性双酯基DA环丙烷;Charette小组[25]报道了铑催化的硝基苯甲酰基重氮与烯烃的不对称环丙烷化反应;Zhang小组[26]发现手性钴卡宾除了可以跟富电子烯烃反应外,也可以与缺电子烯烃反应,合成结构丰富多样的手性环丙烷,其中可能经历了自由基中间体历程;Müller等[27]首次报道了手性铑催化的芳基碘叶立德与末端烯烃的不对称环丙烷化反应,实现了手性双酯基DA环丙烷对映选择性合成;Charette小组[28]发展了铜/苯基双噁唑啉催化的,硝基乙酸酯现场产生碘叶立德与烯烃的不对称环丙烷化反应。另一方面,人们发展了许多高效的合成策略和催化体系,用于DA环丙烷的开环/环化反应。例如,Kerr等[29]报道了首例三氟甲磺酸镱催化的高压下DA环丙烷与吲哚的开环反应;Sibi等[30]发展了首例硝酮与环丙烷的不对称[3+3]环加成反应;Johnson等[31-32]报道了DA环丙烷分别与醛和亚胺发生[3+2]开环/环化反应,他们发现该反应经历了环丙烷的动态动力学拆分历程;Trost等[33]利用手性钯催化剂实现了烯基环丙烷与亚烷基吖内酯的动态动力学不对称[3+2]环加成反应;Waser等[34-35]发展了氨基环丙烷的动态动力学[3+2]开环/环化反应;Maruoka等[36]利用有机小分子硫自由基催化剂,实现了烯基环丙烷与烯基醚的开环/环化反应;冯小明等[37-40]报道了环丙基酮化合物与伯胺的对映选择性开环/环化反应。本文将针对DA环丙烷不对称合成、开环反应、环化反应以及消旋体DA环丙烷的动力学拆分,综述本课题组[41-42]近期利用金属铜和镍与边臂修饰的手性噁唑啉配体,原位生成的手性金属配合物作为催化剂,所取得的一些研究进展。
具有光学活性的环丙烷是非常重要结构单元,广泛存在于天然产物、药物和农药分子中。在一定的条件下,对于含有缺电子芳基和烷基推电子取代基的手性DA环丙烷来说,当其发生开环反应的时候,环丙烷不会发生外消旋化[31, 46, 60]。因此,人们针对不对称环丙烷化反应,发展了各种催化体系,用于合成具有光学活性的环丙烷[1-7]。在这些已经报道的方法中,利用金属卡宾与烯烃发生不对称环丙烷化反应是合成手性DA环丙烷最高效、最实用的方法之一。
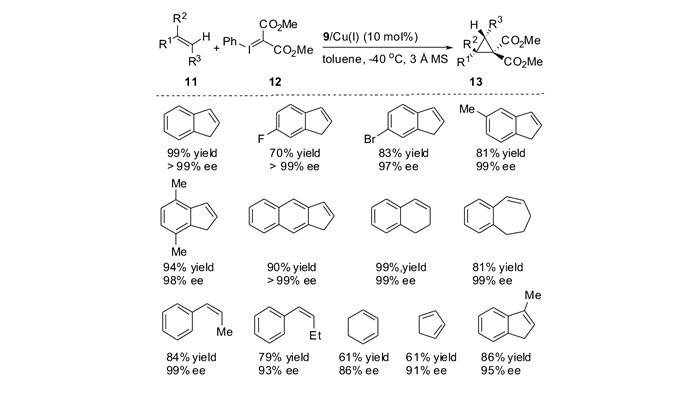
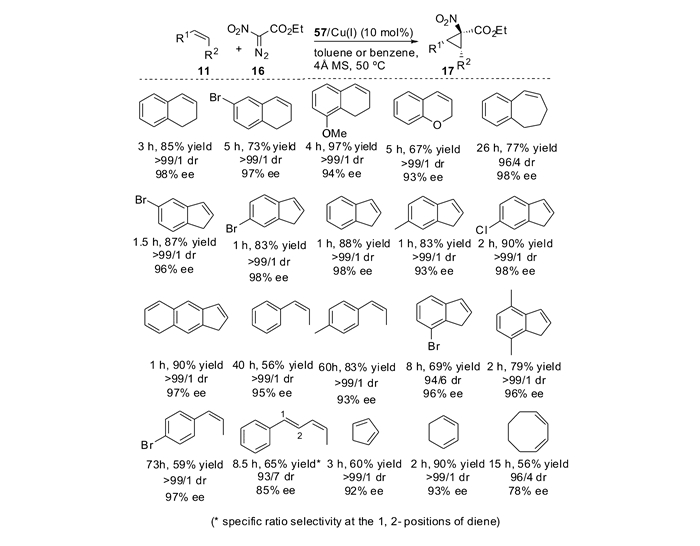
自从Nozaki等[15-16]报道了第1例烯烃与重氮化合物的不对称环丙烷化反应,到目前为止,对于各种各样的末端烯烃,人们已经建立了行之有效的不对称合成环丙烷的方法。然而对于内烯烃,由于金属卡宾对位阻的敏感性,导致其不对称环丙烷化反应往往活性不高、选择性难于控制。因此,亟待于发展更为高效的催化体系,用于实现内烯烃的不对称环丙烷化反应。2012年,本课题组[43]发展了边臂修饰的手性噁唑啉/铜(SaBOX/Cu(Ⅰ))催化体系,实现了基于1, 2-二取代烯烃的不对称环丙烷化反应(图 4)。在温和的反应条件下,无论是顺式-还是反式-1, 2, -二取代烯烃都能够高效地转化为相应的1, 2, 3-三取代DA环丙烷,并且取得了优秀的非对映选择性和对映选择性。
如表 1所示[43],对于一系列苯环上含有推电子或者拉电子取代基的顺式-β-甲基苯乙烯底物,不对称环丙烷化反应可以顺利进行,以72%~84%产率,95/5~96/4 dr(非对映比)和92%~94%ee得到手性DA环丙烷(表 1, Entries 1~4)。二氢萘和吲哚衍生的烯烃也同样适用于该催化体系(Entries 6~7)。该催化体系对于空间位阻更大的反式-β-乙基苯乙烯也同样适用,能够以93/7 dr和86%ee得到相应的DA环丙烷(Entry 8)。
 下载:
导出CSV
下载:
导出CSV

|
||||
| Entry | Alkene | Yield/% | dr | ee/% |
| 1 |  |
84 | 96/4 | 92 |
| 2 |  |
78 | 96/4 | 93 |
| 3 |  |
72 | 95/5 | 94 |
| 4 |  |
72 | 95/5 | 93 |
| 5 |  |
60 | 97/3 | 89 |
| 6 |  |
95 | 97/3 | 89 |
| 7 |  |
60 | 97/3 | 86 |
| 8 |  |
66 | 93/7 | 86 |
另一方面,在2/CuOTf(三氟甲磺酸亚铜)的催化下,具有光学活性的1, 2, 3-三取代DA环丙烷可以通过各种各样的反式-1, 2-二取代烯烃与铜卡宾发生不对称环丙烷化获得(表 2[43])。对于一系列不同的反式-β-甲基苯乙烯衍生物,取代基的位阻和电性对反应的影响较小,均能够取得非对映选择性单一、对映选择性优秀的结果(94%~97%ee)。位阻更大的反式烯烃和三取代烯烃也同样适用,能够以>99/1 dr和96%~98%的ee值获得目标环丙烷化合物(Entries 8~10)。值得一提的是,反式-β-甲基苯乙烯的环丙烷化反应可以放大到50 mmol的规模,在摩尔分数0.05%的催化剂载量下,仍能以44%的产率和98%的对映体过量得到6.16 g手性环丙烷化合物。这是目前以铜作为催化剂发生烯烃与重氮化合物环丙烷化反应的最低催化剂载量。
下载:
导出CSV

|
||||
| Entry | Alkene | Yield/% | dr | ee/% |
| 1 |  |
89 | >99/1 | 96 |
| 2 |  |
99 | >99/1 | 96 |
| 3 |  |
96 | >99/1 | 94 |
| 4 |  |
96 | >99/1 | 97 |
| 5 |  |
73 | >99/1 | 96 |
| 6 |  |
60 | >99/1 | 96 |
| 7 |  |
97 | 93/7 | 96 |
| 8 |  |
64 | >99/1 | 98 |
| 9 |  |
84 | >99/1 | 97 |
| 10 |  |
82 | >99/1 | 96 |
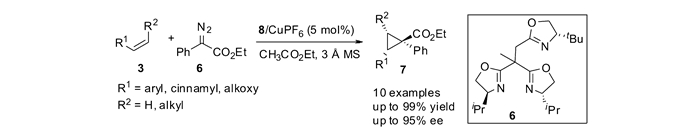
相较于环丙烷化中常用的单取代重氮乙酸酯类试剂,利用重氮化合物中含有推电子和拉电子两个取代基的重氮试剂,例如苯基重氮乙酸酯6,发生环丙烷化反应,可以合成出更具官能团化的1, 1-二取代DA环丙烷。在催化剂8/Cu(Ⅰ)的作用下,烯烃与苯基重氮乙酸酯能够发生不对称环丙烷化反应,以优秀的产率、非对映选择性以及对映选择性高效地合成三取代或四取代的手性环丙烷化合物,见图 5[44]。
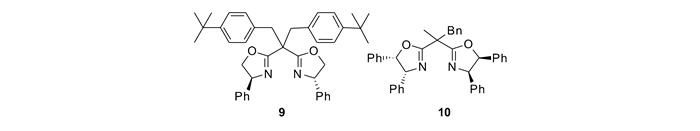
利用烯烃与丙二酸酯衍生的金属卡宾发生不对称催化环丙烷化反应是构建手性1, 1-双酯基环丙烷的最直接的途径之一。然而,仅有少数的铑催化的末端烯烃环丙烷化的例子取得了较高的立体选择性[24, 27]。本课题组[45]发展了双边臂修饰的手性噁唑啉(SaBOX)/铜(9/Cu(Ⅰ))催化体系,实现了内烯烃与丙二酸酯衍生的碘叶立德的不对称环丙烷化反应(见图 6)。
研究发现,反应具有良好的底物普适性,对于各种各样的二取代-、三取代-内烯烃反应均能够高效地发生,以优秀的产率(高达99%)、专一的非对映选择性以及优秀的对映选择性(高达99%ee),获得相应的手性1, 1-二酯基环丙烷化合物(见图 7)[45]。
末端烯烃也同样适用于上述催化体系。如表 3所示[45],无论R1为在邻位、间位还是对位上带有各种不同电子特性取代基的芳基,反应均能够顺利地发生,并取得较高的产率和良好到优秀的对映选择性(87%~96%ee)。
下载:
导出CSV

|
|||
| Entry | R1 | Yield/% | ee/% |
| 1 | p-BrC6H4 | 99 | 95 |
| 2 | Ph | 85 | 91 |
| 3 | p-ClC6H4 | 93 | 95 |
| 4 | p-CF3C6H4 | 99 | 96 |
| 5 | p-ClCH2C6H4 | 79 | 94 |
| 6 | p-MeC6H4 | 99 | 92 |
| 7 | o-MeC6H4 | 99 | 92 |
| 8 | m-MeC6H4 | 97 | 93 |
| 9 | p-PhC6H4 | 95 | 92 |
上述方法原料易得,催化剂廉价,并且反应的立体选择性优异,因此在有机合成中具有很好的实用性。
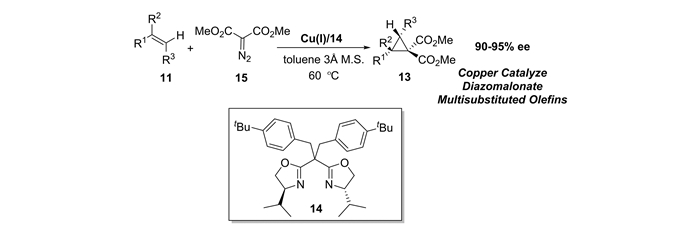
本课题组[46]还研究了由丙二酸酯衍生重氮化合物原位生成丙二酸酯铜卡宾,与多取代烯烃的不对称环丙烷化反应。发现在双边臂取代的噁唑啉/铜(Ⅰ)配合物Cu(Ⅰ)/14催化下,对非末端烯烃可以以高达95%的产率和90%~95%ee获得目标1, 1-双酯基环丙烷类化合物(见图 8)。
硝基环丙烷羧酸酯,作为具有生物活性的α-氨基酸环丙烷的前体,是一类重要的化合物。利用烯烃与硝基金属卡宾发生不对称环丙烷化来合成这类化合物是最高效的方法之一。与其它含有两个拉电子取代基的重氮化合物类似,硝基重氮乙酸酯生成金属卡宾的活性不高。在过去的研究中,当使用铜催化剂分解这类重氮时,往往需要苛刻的反应条件,并且环丙烷化反应的产率和立体选择性均并不理想。本课题组利用边臂修饰的手性噁唑啉配体与铜生成配合物(10/copper(Ⅰ))催化剂(图 6[47]),发展了1, 2-二取代烯烃与α-硝基重氮乙酸酯的不对称环丙烷化反应(图 9[47])。反应具有很好的底物普适性,对于各种各样的顺式-二取代烯烃都能够顺利的发生,能够以56%~97%的产率,94/6~99/1 dr和92%~98%ee得到相应的手性环丙烷。此方法为合成具有光学活性的α-氨基酸环丙烷以及手性非天然氨基酸衍生物提供了简便的途径。
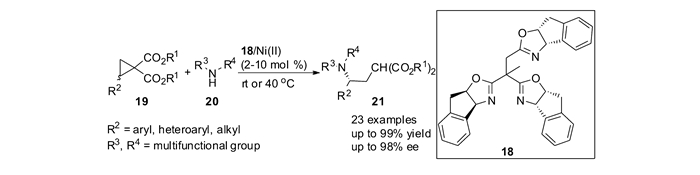
2012年,本课题组[48-49]首次通过仲胺对偕二酯基环丙烷的不对称开环反应,合成了手性γ-芳基-γ-氨基酸衍生物。在18/Ni(Ⅱ)催化下,开环产物21能够取得高达99%的分离产率和98%ee。该反应条件温和,底物范围很广,可以适用于各种芳基、烯基取代的环丙烷19以及芳基和脂肪仲胺20(图 10)。当使用双齿配位的双噁唑配体以及平面三齿配位的双噁唑配体/Ni催化反应时,反应进行的缓慢甚至不反应,对映体过量最高也只有58%。而配体18对该反应具有明显的加速作用和优秀的手性诱导作用。该不对称催化反应为合成手性γ-氨基酸衍生物提供了有效途径。产物经简单转化便可得到多官能团化的手性哌啶衍生物和手性γ-内酰胺。
这一方法学研究还为实现手性环丙烷的动力学拆分提供了高效的方法。如表 4所示[48-49],该方法不仅可以获得90%~97%对映体过量的开环产物21,同时还能够以理想的回收产率拆分得到88%~95%对映体过量的具有光学活性的环丙烷(R)-19。
下载:
导出CSV

|
||||||
| Entry | R2 | Conv. | (R)-19 | 21 | ||
| Yield/% | ee/% | Yield/% | ee/% | |||
| 1 | Ph | 57 | 42 | 93 | 39 | 90 |
| 2 | p-ClC6H4 | 55 | 43 | 95 | 40 | 94 |
| 3 | p-BrC6H4 | 55 | 46 | 93 | 40 | 97 |
| 4 | p-MeC6H4 | 50 | 49 | 88 | 46 | 92 |
| 5 | m-MeC6H4 | 57 | 41 | 93 | 42 | 96 |
本课题组基于仲胺对DA环丙烷的不对称开环研究,将此方法拓展到了醇类和水作为氧原子亲核试剂,用于合成手性γ-羟基酸及其衍生物。γ-羟基酸及其衍生物是一类重要的天然产物以及合成中间体。例如,γ-羟基丁酸(GHB)是一种神经药物,自20世纪60年代开始被用作常用麻醉剂,治疗失眠、抑郁症等。此外,γ-羟基酸及其衍生物也是合成取代的五元内酯的重要中间体。利用醇类化合物或者水作为亲核试剂对DA环丙烷进行开环具有以下难点:1)与氮原子和碳原子亲核试剂相比,氧原子的亲核性相对较弱;2)氧原子亲核试剂和手性配体可能发生与Lewis酸配位的竞争,从而对DA环丙烷发生开环反应的反应活性和对映选择性产生不良影响;3)与仲胺亲核试剂相比较,醇类化合物作为亲核试剂,其立体位阻往往较小,不利于反应的对映选择性控制。特别是以分子体积非常小的水作为亲核试剂时,反应的立体选择性控制的难度会更大。
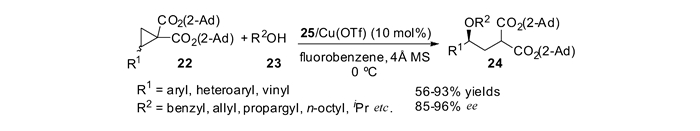
本课题组[50]首先实现了醇类化合物23对偕二酯基DA环丙烷22的不对称开环反应,合成了一系列具有光学活性的γ-羟基酸衍生物。在25/Cu(Ⅱ)的催化下,以氟苯作溶剂,反应能够取得高达93%的产率和96%的对映选择性。该反应底物普适性广,对于多种不同类型的醇类化合物,例如苄基醇、烯丙基醇、炔丙基醇以及多种一级和二级脂肪醇,均能够取得理想的产率和良好到优秀的对映选择性;对于含有不同取代基的DA环丙烷,也同样能够在温和的反应条件下取得理想的结果。开环产物经简单转化可以合成手性的六氢吡喃衍生物以及γ-羟基酸酯见图 11。
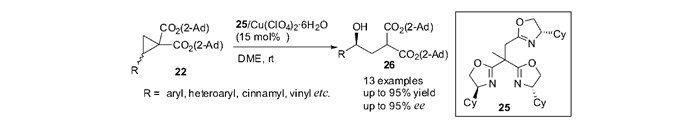
在实现了醇类化合物对DA环丙烷不对称开环反应之后,本课题组[50]尝试利用水对环丙烷22直接进行不对称开环。研究发现,在反应体系中,水的浓度对于环丙烷开环反应的活性影响至关重要:过多的水会导致催化剂失活,从而影响反应活性;而水的浓度过低则会降低目标反应的速率,从而增加体系中的副反应。本课题组采用高氯酸铜水合物既作为Lewis酸催化剂也作为体系中水的存储器,利用其缓释作用调节催化体系中水的浓度,最终实现了水对环丙烷的高活性对映选择性开环。该反应条件温和,在室温下就能够顺利进行。底物普适性广,对于多种不同的基团,例如芳基、杂环芳基、肉桂基和乙烯基取代的DA环丙烷,开环反应均够高效发生,从而以70%~95%产率和82%~95%对映体过量得到开环产物26(图 12)。该反应为高效合成具有光学活性的一系列γ-羟基酸衍生物提供了新的方法。
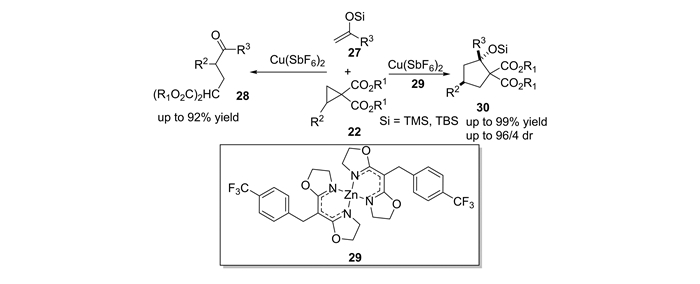
2008年,王忠文小组[51]首次报道了2-取代环丙烷-1, 1-二酯与烯醇硅醚的开环反应。2009年,本课题组[52]发展了DA环丙烷与烯醇硅醚的反应,通过配体调节催化剂Lewis酸的酸性,实现了开环反应和[3+2]环化反应之间的调控。当使用Cu(SbF6)2为催化剂时,烯醇硅醚27对DA环丙烷22发生亲核开环反应,以高达92%的产率得到非环状的1, 6二羰基化合物28;当使用Cu(SbF6)2与锌配合物29为催化剂时,DA环丙烷22与烯醇硅醚27能够发生[3+2]环化反应,以高达99%的产率和高达96/4的非对映选择性得到多官能团化的环戊烷产物30(图 13)。有趣的是,研究发现锌配合物29不能够催化环丙烷的[3+2]环化反应和开环反应。
桥碳上含有羟基官能团的双环[n.3.0]化合物是一大批具有生物生物活性天然产物的核心骨架。DA环丁烷和环状烯醇硅醚的[3+2]环化反应为构建官能团化的环戊烷并环结构提供了高效的方法。本课题组[53]运用廉价的金属铜配合物为催化剂,以高达92%的产率和优秀的非对映选择性合成了一系列带有多个连续立体中心的、具有[n.3.0]双环结构的化合物32(图 14)。值得注意的是,研究发现环丙烷上大位阻的酯基取代基对于反应的非对映选择性控制十分关键。当使用结构刚性的大位阻2-金刚烷酯基时,可以得到最好的结果。通过对照实验以及与北京大学余志祥教授合作进行计算,阐明反应的高立体选择性产生于反应中分子内关环一步。采用大位阻基团取代的环丙烷酯基以及硅基,均会增加环化过渡态的能量。这一计算结果与实验现象相符。
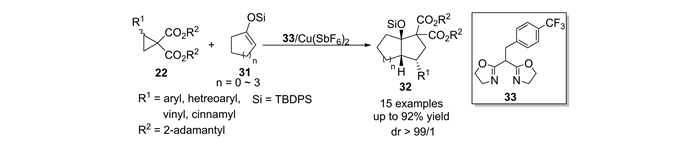
本课题组[54]接下来开展了上述反应的不对称催化研究。在手性铜催化剂34的作用下,合成了一系列具有光学活性的3α-羟基双碳环[n.3.0]化合物(图 15)。从环酮化合物衍生而来的具有5~7元环的烯醇硅醚底物也被应用于与DA环丙烷的不对称[3+2]环化反应中,并取得了理想的产率、>99/1的非对映选择性以及高达99%ee。此外,苯并环酮衍生的烯醇硅醚也能顺利反应,能够以80%~98%的产率、>99/1 dr和91%~94%ee。这是关于DA环丙烷与烯醇硅醚的不对称催化[3+2]环化反应的首例报道。
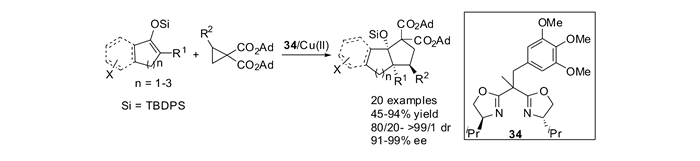
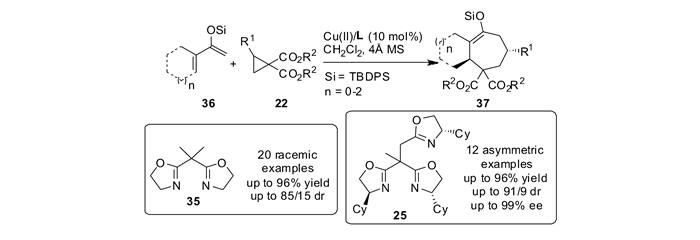
DA环丙烷与烯醇硅醚的共轭二烯反应,在前人的报道[51, 56]中往往只能得到开环产物或者[3+2]环化产物。本课题组[55]发现双噁唑啉35和金属铜盐可以催化DA环丙烷22和烯醇硅醚共轭双烯36的[4+3]环化反应。利用共轭的烯醇硅醚作为双烯体,实现了Lewis酸催化的其与推-拉电子环丙烷的[4+3]环加成反应,为多官能团化的七元环化合物37的合成提供了一种新的途径。该方法具有广谱的底物范围,适用于单环七元环的合成,以及[n, 5, 0]并环化合物的合成,同时还能合成[3.3.2]桥环化合物,取得了最高98%的分离产率和最高85:15的dr值。本课题组以边臂修饰的手性噁唑啉为配体,进一步研究了铜催化的双烯体与推-拉电子环丙烷的不对称[4+3]环加成反应。通过对配体边臂进行了详细的研究,发现三噁唑啉配体25能够明显地提高反应活性,并具有更好的手性诱导效果。该反应的底物普适性好,对于环状烯烃和开链烯烃均能顺利进行,取得高达96%的产率和高达99%的对映选择性(见图 16)。
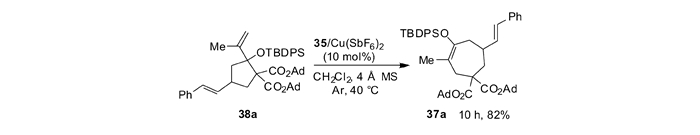
通过1H NMR跟踪实验对机理进行了研究,发现反应是分步进行的,首先生成[3+2]环化产物,经过开环后环化到热力学稳定的[4+3]环化产物。根据上述现象,本课题组[55]设计了对照实验。发现[3+2]环化产物38a在噁唑啉/铜配合物的催化作用下,能够以82%的产率转化为[4+3]环化产物37a(见图 17)。
吲哚及其类似物是许多具有重要生理活性的生物碱都含有的结构单元。吲哚的3位碳原子具有亲核性,在Lewis酸的作用下,能够对DA环丙烷发起亲核进攻,得到环丙烷开环产物[57-58]。本课题组[59]利用边臂修饰的手性噁唑啉配体41与二价金属铜盐原位生成的手性Lewis酸为催化剂,实现了吲哚与DA环丙烷的不对称[3+2]环化反应。对于含有多种不同取代基的吲哚类化合物以及不同的DA环丙烷,反应都能够顺利进行,从而以理想的产率得到具有优秀的非对映选择性(高达>50/1 dr)和对映选择性(高达96%ee)的环戊烷化产物(见图 18)。
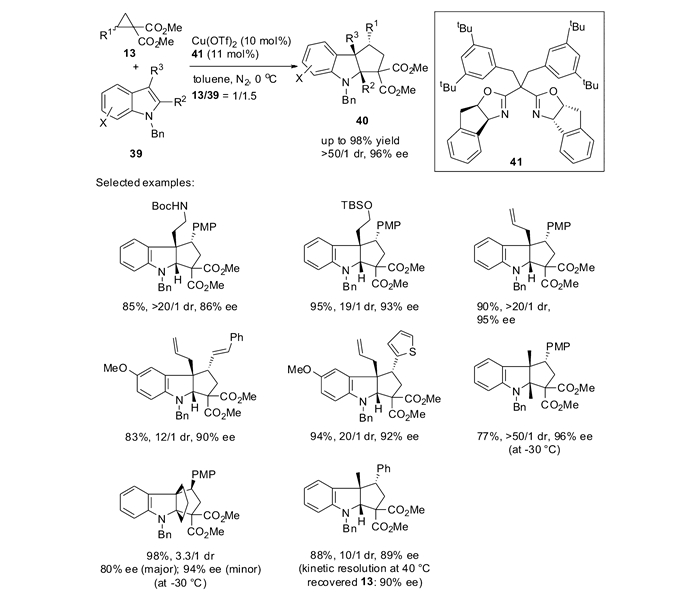
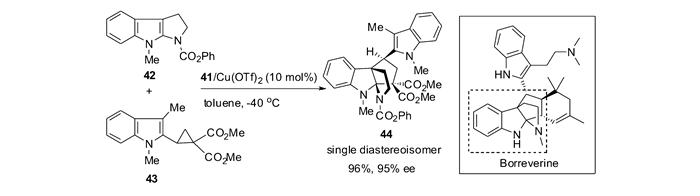
应用上述反应方法学,以吡咯并吲哚42为底物与2-吲哚DA环丙烷反应,可以96%产率,>99/1 dr和95%ee方便的构建天然产物borreverine的四环核心骨架(图 19)[59]。
有趣的是,当使用2-甲基取代吲哚45时,未检测到[3+2]环化产物,只得到环丙烷开环的产物46,产率为80%,对映体过量值为92%ee(图 20) [59]。
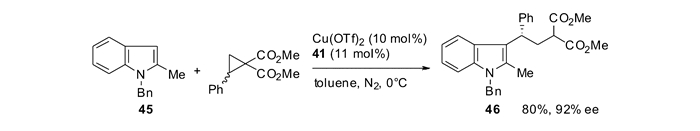
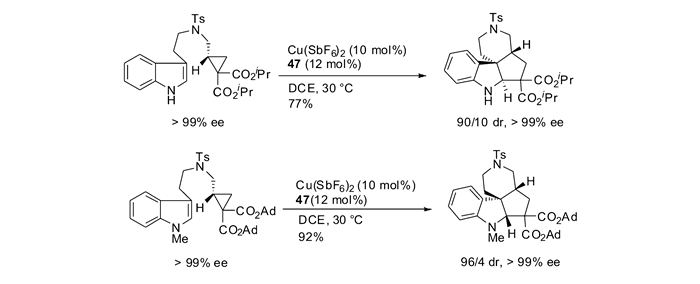
本课题组[60]还研究了DA环丙烷和吲哚的分子内立体选择性[3+2]环化反应,在配合物Cu(Ⅱ)/47的催化作用下,构建了一系列四环螺吲哚啉骨架。研究发现DA环丙烷上的酯基取代基能够对反应的立体选择性实现调控(表 5)。通过选择不同的酯基,能够以优秀的非对映选择性分别得到49(trans产物)和50(cis产物)两种不同的螺吲哚啉化合物。当环丙烷上酯基为异丙酯时,可以以最高77%的分离产率和>90/10的非对映选择性得到trans产物(Entry 4);当环丙烷上酯基为2-金刚烷酯时以最高79%的分离产率和最高5/95的非对映选择性得到cis产物(Entry 13)。当使用光学纯的原料时,可以得到手性完全保留的具有光学活性的螺环吲哚啉产物,并且反应的非对映选择性保持不变(图 21)[60]。为了更深入的理解反应选择性产生的原因,本课题组和Houk小组合作,对反应过程进行了DFT计算。通过计算发现,由于异丙酯和吲哚附芳香环之间有微弱的吸引作用,使得TS1-trans能量较低,有利于trans产物的生成;而当使用2-金刚烷酯时,由于位阻原因,使得TS1-trans能量较高,有利于cis产物的生成。
 下载:
导出CSV
下载:
导出CSV

|
||||
| Entry | R1 | R2 | Yield/% | dr(49/50) |
| 1 | H | Et | 83 | 83/17 |
| 2 | H | Me | 87 | 74/26 |
| 3 | H | nHex | 77 | 78/22 |
| 4 | H | iPr | 77 | 90/10 |
| 5 | H | cHex | 83 | 88/12 |
| 6 | H | 3-Pent | 45 | 89/11 |
| 7 | H | tBu | trace | - |
| 8 | H | 1-Ad | trace | - |
| 9 | H | CH2tBu | 64 | 15/85 |
| 10 | H | CH2-1-Ad | 52 | 9/91 |
| 11 | H | 2-Ad | 61 | 6/94 |
| 12 | CH3 | iPr | 85 | 84/16 |
| 13 | CH3 | 2-Ad | 79 | 5/95 |
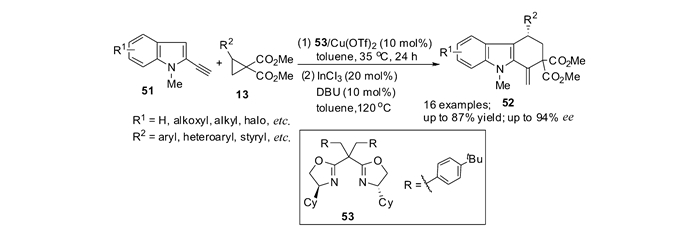
Kerr等[62-63]利用吲哚与DA环丙烷先发生开环反应再发生Conia-ene环化反应,发展了一种简洁高效地合成四氢咔唑类化合物的方法。最近,本课题组[61]实现了DA环丙烷13与2-炔基吲哚51的对映选择性[3+3]环化反应,“一锅法”合成了具有光学活性的1, 2, 3, 4-四氢咔唑化合物52。在手性Lewis酸催化剂53/Cu(OTf)2的作用下,吲哚对DA环丙烷的亲核开环能够顺利进行,继而以InCl3作为Lewis酸,在加热条件下发生Conia-ene环化反应。通过对溶剂、金属、配体、DA环丙烷酯基大小及吲哚氮上取代基的类型的研究,最终能够以高达87%的产率和93%ee得到目标产物。该反应原料简单易得,具有良好的底物普适性(图 22)。
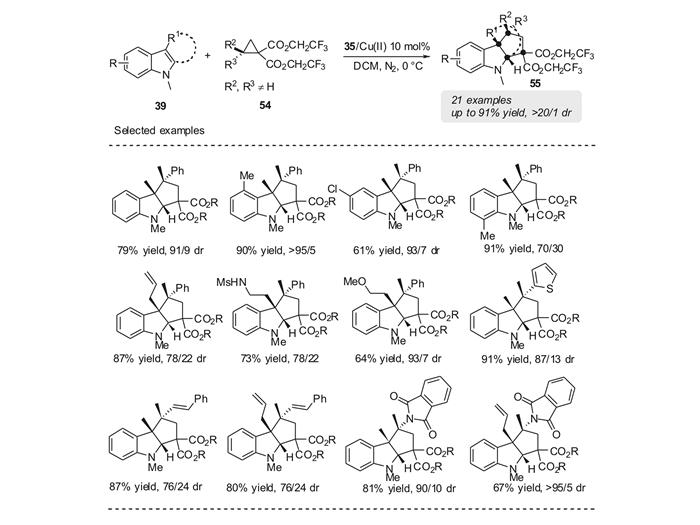
对于推电子基团位点含有两个取代基的DA环丙烷(四取代DA环丙烷),目前也有一些研究的报道[64-71]。当该类DA环丙烷发生亲核开环反应时, 能够产生新的季碳中心。在Cu(Ⅱ)/35为催化剂的条件下,本课题组[72]首次实现了吲哚与四取代DA环丙烷54的[3+2]环加成反应。该反应条件温和,底物普适性好。对芳环上含有不同取代基的吲哚、色醇、色胺均能以优秀的产率和中等到优秀的非对映选择性(70/30~>95/5 dr)得到四氢吲哚的衍生物55。对于噻吩基、呋喃基、肉桂基、邻苯二甲酰胺基取代的DA环丙烷,反应也均能够顺利的进行,取得优秀的产率和中等到优秀的非对映选择性(76/24~>95/5 dr)。用该策略可以高效构建吲哚并五元碳环结构,能够在五元环上一次性地引入3个甚至4个连续的季碳中心(图 23)。
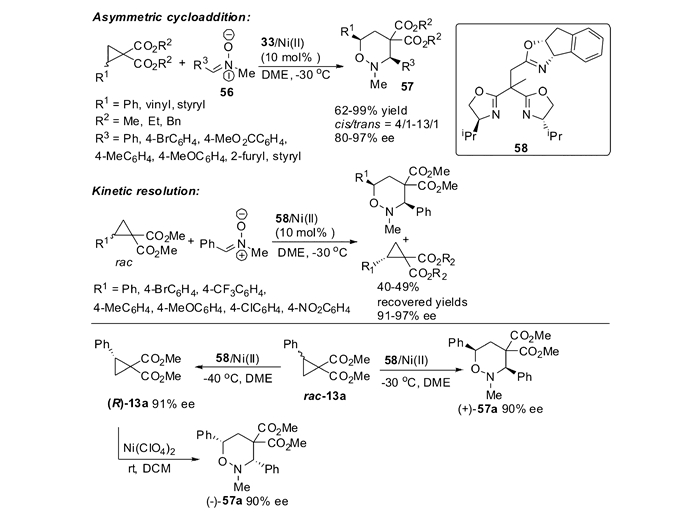
硝酮、氮杂亚胺叶立德等1, 3偶极子均能与DA环丙烷发生1, 3-偶极环加成反应。本课题组致力于发展DA环丙烷与1, 3偶极子的不对称环加成反应,用于构建多种具有高合成价值的杂环化合物。在本课题组的研究过程中,Sibi等[30]于2005年使用手性镍催化剂,简洁高效地实现了环丙烷与硝酮的首次不对称[3+3]环加成反应。当他们使用含有拉电子活化基团的环丙烷时,能够以高达99%的产率和95%的对映体过量值获得[3+3]环加成产物。然而,当他们使用DA环丙烷时,尽管反应仍然可以得到优秀的产率和对映选择性,反应的非对映选择性却并不理想(0.8/1~1.4/1 dr)。2007年,本课题组[73]发展了一种手性假C3对称的三噁唑啉/镍(Ⅱ)催化剂体系,用于催化2-取代环丙烷-1, 1-二酯类化合物与硝酮56的环加成反应,最高可获得93/7 dr和80%~97%的对映体过量值(图 24)。该催化体系还能用于DA环丙烷的动力学拆分,获得具有优秀的对映体过量值的手性环丙烷化合物。本课题组通过上述发展的不对称[3+3]环加成反应,以及环丙烷的动力学拆分/环加成串联反应,为简洁高效地合成手性四氢1, 2-噁嗪的两种对映异构体提供了有效的方法。
二氢和四氢异喹啉(喹啉)是一类重要的合成中间体,这些结构单元广泛存在于生物碱和其他类型的生物活性分子中。利用手性In-TOX(18)/Ni(Ⅱ)配合物催化剂首次实现了氮杂亚胺叶立德59与偕二酯基DA环丙烷的不对称催化的[3+3]环化反应,可以最高99%的产率、高达95/5 dr和97%的对映体过量获得新颖的6, 6, 6-三环二氢异喹啉衍生物,并且产物在还原剂NaBH3CN的作用下可以方便地转化成四氢异喹啉衍生物60(图 25)[74]。
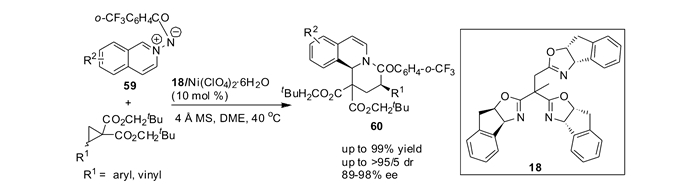
在In-TOX(18)/Ni(Ⅱ)的催化下,本课题组[74]研究了在最佳的反应条件下,不同的芳香氮杂亚胺叶立德底物与对甲氧基苯基取代的双酯基环丙烷的[3+3]环化反应。如表 6所示,异喹啉环上甲基的位置对反应的立体选择性略有影响,当使用底物59b和59c的时候,反应均能以理想的产率、优秀的非对映选择性和对映选择性得到目标产物(产率85%~94%、92/8~>95/5 dr和86%~96%ee, 表 6, Entries 2~3)。此外,该催化体系对于喹啉类底物59e~59f也能够适用,尽管反应的非对映选择性不够理想,仍然能够以优秀的产率和对映选择性得到[3+3]环化产物(表 6, Entries 5~6)。
下载:
导出CSV

|
|||||
| Entry | 59 | Time/h | Yield/% | dr(cis/trans) | ee/% |
| 1 | 59a | 7 | 91 | >95/5 | 94 |
| 2 | 59b | 9 | 94 | >95/5 | 96 |
| 3 | 59c | 9 | 85 | 92/8 | 86 |
| 4 | 59d | 9 | 81 | 75/25 | 88/86 |
| 5 | 59e | 5.5 | 92 | 42/58 | 90/95 |
| 6 | 59f | 14 | 99 | 58/42 | 88/91 |
在该反应中,本课题组[74]发现手性三噁唑啉配体18的边臂对于取得高对映选择性是至关重要的。在氮杂亚胺叶立德上引入三氟甲基能够加快反应速度和提高反应的对映选择性。研究发现,对于苯基以及相对缺电子芳环取代的偕二酯基DA环丙烷的[3+3]环化反应是不对称动力学拆分过程;而对于富电子芳环取代的偕二酯基DA环丙烷存在不对称动态动力学过程。通过对该反应过程和立体诱导模型进行了详细的DFT理论计算,解释了高立体选择性的成因和配体边臂的作用。
在过去的10年间,本课题组发展了一系列DA环丙烷的不对称合成方法以及不对称开环、环化方法。利用手性铜(Ⅰ)与重氮化合物、碘叶立德等卡宾前体原位生成金属铜卡宾物种与多取代烯烃发生不对称环丙烷化反应;利用手性铜(Ⅱ)、镍(Ⅱ)催化剂,发展了胺、醇、水、硝酮、氮杂亚胺叶立德、烯醇硅醚和吲哚等各种各样的亲核试剂对DA环丙烷发生立体选择性开环、环化反应。这些方法为构建结构多样性的有用的手性骨架,例如具有光学活性的α-氨基酸环丙烷、γ-官能团化羰基化合物以及具有多个手性中心的杂环化合物等。然而,在DA环丙烷的成环、开环、环化领域仍然存在这一些尚未解决的挑战。例如,四取代烯烃环丙烷化反应的活性和立体选择性控制仍然是一个挑战;当DA环丙烷的推电子取代基为烷基时,其不对称开环反应仍然较为困难;对于含有一个拉电子取代基的DA环丙烷,利用Lewis催化的不对称开环反应仍然未能有效解决[75]。与此同时,DA环丙烷发生动态动力学拆分途径的不对称开环反应仍然局限于富电子的取代基,例如对甲氧基苯基、烷氧基等。上述问题需要化学家们发展新型的催化剂体系和新的反应策略来解决。此外,本课题组期望本文所综述的不对称催化反应能够更为广泛地应用于天然产物全合成以及重要手性药物分子的合成中。
Danishefsky S. Electrophilic Cyclopropanes in Organic Synthesis[J]. Acc Chem Res, 1979, 12(2): 66-72. doi: 10.1021/ar50134a004
Reissig H U. Donor-Acceptor-Substituted Cyclopropanes-Versatile Building-Blocks in Organic-Synthesis[J]. Top Curr Chem, 1988, 144: 73-135. doi: 10.1007/BFb0111225
Yu M, Pagenkopf B L. Recent Advances in Donor-Acceptor(DA) Cyclopropanes[J]. Tetrahedron, 2005, 61(2): 321-347. doi: 10.1016/j.tet.2004.10.077
Waser J, De Simone F. Cyclization and Cycloaddition Reactions of Cyclopropyl Carbonyls and Imines[J]. Synthesis, 2009, (20): 3353-3374.
Mel'nikov M Y, Budynina E M, Ivanova O A. Recent Advances in Ring-Forming Reactions of Donor Acceptor Cyclopropanes[J]. Mendeleev Commun, 2011, 21(6): 293-301. doi: 10.1016/j.mencom.2011.11.001
Schneider T F, Kaschel J, Werz D B. A New Golden Age for Donor-Acceptor Cyclopropanes[J]. Angew Chem Int Ed, 2014, 53(22): 5504-5523. doi: 10.1002/anie.v53.22
Grover H K, Emmett M R, Kerr M A. Carbocycles from Donor-Acceptor Cyclopropanes[J]. Org Biomol Chem, 2015, 13(3): 655-671. doi: 10.1039/C4OB02117G
Lebel H, Marcoux J F, Molinaro C. Stereoselective Cyclopropanation Reactions[J]. Chem Rev, 2003, 103(4): 977-1050. doi: 10.1021/cr010007e
Muller P. Asymmetric Transfer of Carbenes with Phenyliodonium Ylides[J]. Acc Chem Res, 2004, 37(4): 243-251. doi: 10.1021/ar0202619
Pellissier H. Recent Developments in Asymmetric Cyclopropanation[J]. Tetrahedron, 2008, 64(30/31): 7041-7095.
Pfaltz A. Comprehensive Asymmetric Catalysis[M]. Jacobsen E N, Pflatz A, Yamamoto H, Eds. Berlin:Springer-Verlag, 1999, Ⅱ:513.
Reissig H U, Zimmer R. Donor-acceptor-substituted Cyclopropane Derivatives and Their Application in Organic Synthesis[J]. Chem Rev, 2003, 103(4): 1151-1196. doi: 10.1021/cr010016n
Carson C A, Kerr M A. Heterocycles from Cyclopropanes:Applications in Natural Product Synthesis[J]. Chem Soc Rev, 2009, 38(11): 3051-3060. doi: 10.1039/b901245c
Reissig H U, Zimmer R. Thrilling Strain-Donor-Acceptor-Substituted Cyclobutanes for the Synthesis of (Hetero)Cyclic Compounds[J]. Angew Chem Int Ed, 2015, 54(17): 5009-5011. doi: 10.1002/anie.v54.17
Nozaki H, Moriuti S, Takaya H. Asymmetric Induction in Carbenoid Reaction by Means of a Dissymmetric Copper Chelate[J]. Tetrahedron Lett, 1966, (43): 5239-5240.
Nozaki H, Takaya H, Moriuti S. Homogeneous Catalysis in Decomposition of Diazo Compounds by Copper Chelates[J]. Tetrahedron, 1968, 24(9): 3655-3656. doi: 10.1016/S0040-4020(01)91998-2
Fritschi H, Leutenegger U, Pfaltz A. Chiral Copper-Semicorrin Complexes as Enantioselective Catalysts for the Cyclopropanation of Olefins by Diazo Compounds[J]. Angew Chem Int Ed, 1986, 25(11): 1005-1006. doi: 10.1002/(ISSN)1521-3773
Fritschi H, Leutenegger U, Siegmann K. Semicorrin Metal Complexes as Enantioselective Catalysts.Part 1.Synthesis of Chiral Semicorrin Ligands and General Concepts[J]. Helv Chim Acta, 1988, 71(6): 1541-1552. doi: 10.1002/(ISSN)1522-2675
Fritschi H, Leutenegger U, Pfaltz A. Semicorrin Metal Complexes as Enantioselective Catalysts.Part 2.Enantioselective Cyclopropane Formation from Olefins with Diazo Compounds Catalyzed by Chiral (Semicorrinato)Copper Complexes[J]. Helv Chim Acta, 1988, 71(6): 1553-1565. doi: 10.1002/(ISSN)1522-2675
Doyle M P, Bagheri V, Wandless T J. Exceptionally High Trans(Anti) Stereoselectivity in Catalytic Cyclopropanation Reactions[J]. J Am Chem Soc, 1990, 112(5): 1906-1912. doi: 10.1021/ja00161a040
Evans D A, Woerpel K A, Hinman M M. Bis(oxazolines) as Chiral Ligands in Metal-Catalyzed Asymmetric Reactions. Catalytic, Asymmetric Cyclopropanation of Olefins[J]. J Am Chem Soc, 1991, 113(2): 726-728. doi: 10.1021/ja00002a080
Ito K, Katsuki T. Asymmetric Cyclopropanation of E-Oleflns Using a Copper Complex of an Optically Active Bipyridine as a Catalyst[J]. Synlett, 1993, (9): 638-640.
Suematsu H, Kanchiku S, Uchida T. Construction of Aryliridium-Salen Complexes:Enantio-and Cis-Selective Cyclopropanation of Conjugated and Nonconjugated Olefins[J]. J Am Chem Soc, 2008, 130(31): 10327-10337. doi: 10.1021/ja802561t
Nishimura T, Maeda Y, Hayashi T. Asymmetric Cyclopropanation of Alkenes with Dimethyl Diazomalonate Catalyzed by Chiral Diene-Rhodium Complexes[J]. Angew Chem Int Ed, 2010, 49(40): 7324-7327. doi: 10.1002/anie.v49:40
Zhu S, Perman J A, Zhang X P. Acceptor/Acceptor-Substituted Diazo Reagents for Carbene Transfers:Cobalt-Catalyzed Asymmetric Z-Cyclopropanation of Alkenes with α-Nitrodiazoacetates[J]. Angew Chem Int Ed, 2008, 47(44): 8460-8463. doi: 10.1002/anie.v47:44
Lindsay V N G, Nicolas C, Charette A B. Asymmetric Rh(Ⅱ)-Catalyzed Cyclopropanation of Alkenes with Diacceptor Diazo Compounds:p-Methoxyphenyl Ketone as a General Stereoselectivity Controlling Group[J]. J Am Chem Soc, 2011, 133(23): 8972-8981. doi: 10.1021/ja201237j
M ller P, Ghanem A. Rh(Ⅱ)-Catalyzed Enantioselective Cyclopropanation of Olefins with Dimethyl Malonate via in Situ Generated Phenyliodonium Ylide[J]. Org Lett, 2004, 6(23): 4347-4350. doi: 10.1021/ol048159u
Moreau B, Charette A B. Expedient Synthesis of Cyclopropane α-Amino Acids by the Catalytic Asymmetric Cyclopropanation of Alkenes Using Iodonium Ylides Derived from Methyl Nitroacetate[J]. J Am Chem Soc, 2005, 127(51): 18014-18015. doi: 10.1021/ja056192l
Harrington P, Kerr M A. The High Pressure Reaction of Cyclopropanes with Indoles Catalyzed by Ytterbium Triflate[J]. Tetrahedron Lett, 1997, 38(34): 5949-5952. doi: 10.1016/S0040-4039(97)01351-8
Sibi M P, Ma Z, Jasperse C P. Enantioselective Addition of Nitrones to Activated Cyclopropanes[J]. J Am Chem Soc, 2005, 127(16): 5764-5765. doi: 10.1021/ja0421497
Parsons A T, Johnson J S. Catalytic Enantioselective Synthesis of Tetrahydrofurans:A Dynamic Kinetic Asymmetric[3+2] Cycloaddition of Racemic Cyclopropanes and Aldehydes[J]. J Am Chem Soc, 2009, 131(9): 3122-3123. doi: 10.1021/ja809873u
Parsons A T, Smith A G, Neel A J. Dynamic Kinetic Asymmetric Synthesis of Substituted Pyrrolidines from Racemic Cyclopropanes and Aldimines:Reaction Development and Mechanistic Insights[J]. J Am Chem Soc, 2010, 132(28): 9688-9692. doi: 10.1021/ja1032277
Trost B M, Morris P J. Palladium-Catalyzed Diastereo-and Enantioselective Synthesis of Substituted Cyclopentanes Through a Dynamic Kinetic Asymmetric Formal[3+2] -Cycloaddition of Vinyl Cyclopropanes and Alkylidene Azlactones[J]. Angew Chem Int Ed, 2011, 50(27): 6167-6170. doi: 10.1002/anie.v50.27
de Nanteuil F, Waser J. Catalytic[3+2] Annulation of Aminocyclopropanes for the Enantiospecific Synthesis of Cyclopentylamines[J]. Angew Chem Int Ed, 2011, 50(50): 12075-12079. doi: 10.1002/anie.v50.50
de Nanteuil F, Serrano E, Perrotta D. Dynamic Kinetic Asymmetric[3+2] Annulation Reactions of Aminocyclopropanes[J]. J Am Chem Soc, 2014, 136(17): 6239-6242. doi: 10.1021/ja5024578
Hashimoto T, Kawamata Y, Maruoka K. An Organic Thiyl Radical Catalyst for Enantioselective Cyclization[J]. Nat Chem, 2014, 6(8): 702-705. doi: 10.1038/nchem.1998
Xia Y, Liu X H, Zheng H F. Asymmetric Synthesis of 2, 3-Dihydropyrroles by Ring-Opening/Cyclization of Cyclopropyl Ketones Using Primary Amines[J]. Angew Chem Int Ed, 2015, 54(1): 227-230. doi: 10.1002/anie.201407880
Xia Y, Lin L, Chang F. Asymmetric Ring-Opening of Cyclopropyl Ketones with Thiol, Alcohol, and Carboxylic Acid Nucleophiles Catalyzed by a Chiral N, N'-Dioxide-Scandium(Ⅲ) Complex[J]. Angew Chem Int Ed, 2015, 54(46): 13952-13956.
Xia Y, Lin L, Chang F. Asymmetric Ring Opening/Cyclization/Retro-Mannich Reaction of Cyclopropyl Ketones with Aryl 1, 2-Diamines for the Synthesis of Benzimidazole Derivatives[J]. Angew Chem Int Ed, 2016, 55(40): 12228-12232. doi: 10.1002/anie.201604735
Fu X, Lin L L, Xia Y. Catalytic Asymmetric[3+3] Annulation of Cyclopropanes with Mercaptoacetaldehyde[J]. Org Biomol Chem, 2016, 14(25): 5914-5917. doi: 10.1039/C6OB00948D
Liao S, Sun X L, Tang Y. Side Arm Strategy for Catalyst Design:Modifying Bisoxazolines for Remote Control of Enantioselection and Related[J]. Acc Chem Res, 2014, 47(8): 2260-2272. doi: 10.1021/ar800104y
Wang L, Tang Y. Asymmetric Ring-Opening Reactions of Donor-Acceptor Cyclopropanes and Cyclobutanes[J]. Isr J Chem, 2016, 56(6/7): 463-475.
Li J, Liao S H, Xiong H. Highly Diastereo-and Enantioselective Cyclopropanation of 1, 2-Disubstituted Alkenes[J]. Angew Chem Int Ed, 2012, 51(35): 8838-8841. doi: 10.1002/anie.v51.35
Xu Z H, Zhu S N, Sun X L. Sidearm Effects in the Enantioselective Cyclopropanation of Alkenes with Aryldiazoacetates Catalyzed by Trisoxazoline/Cu(Ⅰ)[J]. Chem Commun, 2007, (19): 1960-1962. doi: 10.1039/b617967c
Deng C, Wang L J, Zhu J. A Chiral Cagelike Copper(Ⅰ) Catalyst for the Highly Enantioselective Synthesis of 1, 1-Cyclopropane Diesters[J]. Angew Chem Int Ed, 2012, 51(46): 11620-11623. doi: 10.1002/anie.201206376
Deng C, Liu H K, Zheng Z B. Copper-Catalyzed Enantioselective Cyclopropanation of Internal Olefins with Diazomalonates[J]. Org Lett, 2017, 19(21): 5717-5719. doi: 10.1021/acs.orglett.7b02694
Feng L W, Wang P, Wang L. Copper(Ⅰ)/SaBOX Catalyzed Highly Diastereo-and Enantio-Selective Cyclopropanation of cis-1, 2-Disubstituted Olefins with α-Nitrodiazoacetates[J]. Sci Bull, 2014, 60(2): 210-215.
Zhou Y Y, Wang L J, Li J. Side-Arm-Promoted Highly Enantioselective Ring-Opening Reactions and Kinetic Resolution of Donor-Acceptor Cyclopropanes with Amines[J]. J Am Chem Soc, 2012, 134(22): 9066-9069. doi: 10.1021/ja302691r
Kang Q, Wang L, Zheng Z. Sidearm as a Control in the Asymmetric Ring Opening Reaction of Donor-Acceptor Cyclopropane[J]. Chinese J Chem, 2014, 32(8): 669-672. doi: 10.1002/cjoc.201400053
Kang Q K, Wang L, Liu Q J. Asymmetric H2O-Nucleophilic Ring Opening of D-A Cyclopropanes:Catalyst Serves as a Source of Water[J]. J Am Chem Soc, 2015, 137(46): 14594-14597. doi: 10.1021/jacs.5b10310
Fang J, Ren J, Wang Z W. Sc(OTf)3-Catalyzed Smooth Tandem[3+2] Cycloaddition/Ring Opening of Donor-Acceptor Cyclopropane 1, 1-Diesters with Enol Silyl Ethers[J]. Tetrahedron Lett, 2008, 49(47): 6659-6662. doi: 10.1016/j.tetlet.2008.09.028
Qu J P, Deng C, Zhou J. Switchable Reactions of Cyclopropanes with Enol Silyl Ethers. Controllable Synthesis of Cyclopentanes and 1, 6-Dicarbonyl Compounds[J]. J Org Chem, 2009, 74(20): 7684-7689. doi: 10.1021/jo901340v
Qu J P, Liang Y, Xu H. Highly Diastereoselective Construction of Fused Carbocycles from Cyclopropane-1, 1-Dicarboxylates and Cyclic Enol Silyl Ethers:Scope, Mechanism, and Origin of Diastereoselectivity[J]. Chem Eur J, 2012, 18(8): 2196-2201. doi: 10.1002/chem.201103495
Xu H, Qu J P, Liao S. Highly Enantioselective[3+2] Annulation of Cyclic Enol Silyl Ethers with Donor-Acceptor Cyclopropanes:Accessing 3a-Hydroxy[n.3.0] Carbobicycles[J]. Angew Chem Int Ed, 2013, 52(14): 4004-4007. doi: 10.1002/anie.v52.14
Xu H, Hu J L, Wang L. Asymmetric Annulation of Donor-Acceptor Cyclopropanes with Dienes[J]. J Am Chem Soc, 2015, 137(25): 8006-8009. doi: 10.1021/jacs.5b04429
Chagarovskiy A O, Budynina E M, Ivanova O A. Lewis Acid-catalyzed Reactions of Donor-Acceptor Cyclopropanes with Furan Derivatives[J]. Tetrahedron, 2009, 65: 5385-5392. doi: 10.1016/j.tet.2009.04.061
Emmett M R, Kerr M A. Nucleophilic Ring Opening of Cyclopropane Hemimalonates Using Internal Bronsted Acid Activation[J]. Org Lett, 2011, 13(16): 4180-4183. doi: 10.1021/ol201486x
Wales S M, Walker M M, Johnson J S. Asymmetric Synthesis of Indole Homo-Michael Adducts via Dynamic Kinetic Friedel-Crafts Alkylation with Cyclopropanes[J]. Org Lett, 2013, 15(10): 2558-2561. doi: 10.1021/ol4010646
Xiong H, Xu H, Liao S. Copper-Catalyzed Highly Enantioselective Cyclopentannulation of Indoles with Donor-Acceptor Cyclopropanes[J]. J Am Chem Soc, 2013, 135(21): 7851-7854. doi: 10.1021/ja4042127
Zhu J, Liang Y, Wang L. Remote Ester Groups Switch Selectivity:Diastereodivergent Synthesis of Tetracyclic Spiroindolines[J]. J Am Chem Soc, 2014, 136(19): 6900-6903. doi: 10.1021/ja503117q
Liu Q J, Yan W G, Wang L. One-Pot Catalytic Asymmetric Synthesis of Tetrahydrocarbazoles[J]. Org Lett, 2015, 17(16): 4014-4017. doi: 10.1021/acs.orglett.5b01909
Grover H K, Lebold T P, Kerr M A. Tandem Cyclopropane Ring-Opening/Conia-ene Reactions of 2-Alkynyl Indoles:A[3+3] Annulative Route to Tetrahydrocarbazoles[J]. Org Lett, 2011, 13(2): 220-223. doi: 10.1021/ol102627e
Talukdar R, Tiwari D P, Saha A. Diastereoselective Synthesis of Functionalized Tetrahydrocarbazoles via a Domino-Ring Opening-Cyclization of Donor-Acceptor Cyclopropanes with Substituted 2-Vinylindoles[J]. Org Lett, 2014, 16(15): 3954-3957. doi: 10.1021/ol501763n
Sherry B D, Furstner A. Iron-catalyzed Addition of Grignard Reagents to Activated Vinyl Cyclopropanes[J]. Chem Commun, 2009, (46): 7116-7118. doi: 10.1039/b918818e
Xing S, Pan W, Liu C. Efficient Construction of Oxa-and Aza-[n.2.1] Skeletons:Lewis Acid Catalyzed Intramolecular[3+2] Cycloaddition of Cyclopropane 1, 1-Diesters with Carbonyls and Imines[J]. Angew Chem Int Ed, 2010, 49(18): 3215-3218. doi: 10.1002/anie.v49:18
Smith A G, Slade M C, Johnson J S. Cyclopropane-Aldehyde Annulations at Quaternary Donor Sites:Stereoselective Access to Highly Substituted Tetrahydrofurans[J]. Org Lett, 2011, 13(8): 1996-1999. doi: 10.1021/ol200395e
Yu Q, Ma S. Lewis Acid-Catalyzed Unexpected Selective C-C Bond Cleavage:An Efficient and Mild Construction of Cyclopentenes[J]. Chem Commun, 2012, 48(96): 11784-11786. doi: 10.1039/c2cc36672j
Wenz D R, Read de Alaniz. Aza-Piancatelli Rearrangement Initiated by Ring Opening of Donor-Acceptor Cyclopropanes[J]. J Org Lett, 2013, 15(13): 3250-3253. doi: 10.1021/ol401248p
Zhu W, Fang J, Liu Y. Lewis Acid Catalyzed Formal Intramolecular[3+2] Cross-Cycloaddition of Cyclopropane 1, 1-Diesters with Alkenes:General and Efficient Strategy for Construction of Bridged[n.2.1] Carbocyclic Skeletons[J]. Angew Chem Int Ed, 2013, 52(7): 2032-2037. doi: 10.1002/anie.v52.7
Haubenreisser S, Hensenne P, Schröder S. The Alkynyl Moiety as a Donor for Donor-Acceptor Cyclopropanes[J]. Org Lett, 2013, 15(9): 2262-2265. doi: 10.1021/ol400809n
Mackay W D, Fistikci M, Carris R M. Lewis Acid Catalyzed (3+2)-Annulations of Donor Acceptor Cyclopropanes and Ynamides[J]. Org Lett, 2014, 16(6): 1626-1629. doi: 10.1021/ol500256n
严文广, 王盼, 王丽佳. 铜(Ⅱ)催化的吲哚与四取代DA环丙烷[3+2]开环/环化反应[J]. 化学学报, 2017,75,(8): 783-787. YAN Wenguang, WANG Pan, WANG Lijia. Copper Catalyzed[3+2] Annulation of Indoles with 1, 1, 2, 2-Tetrasubstituted Donor-Acceptor Cyclopropanes[J]. Acta Chim Sin, 2017, 75(8): 783-787.
Kang Y B, Sun X L, Tang Y. Highly Enantioselective and Diastereoselective Cycloaddition of Cyclopropanes with Nitrones and Its Application in the Kinetic Resolution of 2-Substituted Cyclopropane-1, 1-Dicarboxylates[J]. Angew Chem Int Ed, 2007, 46(21): 3918-3921. doi: 10.1002/(ISSN)1521-3773
Zhou Y Y, Li J, Ling L. Highly Enantioselective[3+3] Cycloaddition of Aromatic Azomethine Imines with Cyclopropanes Directed by π-π Stacking Interactions[J]. Angew Chem Int Ed, 2013, 52(5): 1452-1456. doi: 10.1002/anie.201207576
Liu Q S, Wang D Y, Yang Z J. Ni-Al Bimetallic Catalyzed Enantioselective Cycloaddition of Cyclopropyl Carboxamide with Alkyne[J]. J Am Chem Soc, 2017, 139(50): 18150-18153. doi: 10.1021/jacs.7b09947

图 1 获得手性环丙烷的对映选择性策略
Figure 1 Enantioselective protocols to obtain enantiopure cyclopropanes
















|
|
||||
| Entry | Alkene | Yield/% | dr | ee/% |
| 1 | |
84 | 96/4 | 92 |
| 2 | |
78 | 96/4 | 93 |
| 3 | |
72 | 95/5 | 94 |
| 4 | |
72 | 95/5 | 93 |
| 5 | |
60 | 97/3 | 89 |
| 6 | |
95 | 97/3 | 89 |
| 7 | |
60 | 97/3 | 86 |
| 8 | |
66 | 93/7 | 86 |
 下载: 导出CSV
下载: 导出CSV
|
|
||||
| Entry | Alkene | Yield/% | dr | ee/% |
| 1 | |
89 | >99/1 | 96 |
| 2 | |
99 | >99/1 | 96 |
| 3 | |
96 | >99/1 | 94 |
| 4 | |
96 | >99/1 | 97 |
| 5 | |
73 | >99/1 | 96 |
| 6 | |
60 | >99/1 | 96 |
| 7 | |
97 | 93/7 | 96 |
| 8 | |
64 | >99/1 | 98 |
| 9 | |
84 | >99/1 | 97 |
| 10 | |
82 | >99/1 | 96 |
下载: 导出CSV
|
|
|||
| Entry | R1 | Yield/% | ee/% |
| 1 | p-BrC6H4 | 99 | 95 |
| 2 | Ph | 85 | 91 |
| 3 | p-ClC6H4 | 93 | 95 |
| 4 | p-CF3C6H4 | 99 | 96 |
| 5 | p-ClCH2C6H4 | 79 | 94 |
| 6 | p-MeC6H4 | 99 | 92 |
| 7 | o-MeC6H4 | 99 | 92 |
| 8 | m-MeC6H4 | 97 | 93 |
| 9 | p-PhC6H4 | 95 | 92 |
下载: 导出CSV
|
|
||||||
| Entry | R2 | Conv. | (R)-19 | 21 | ||
| Yield/% | ee/% | Yield/% | ee/% | |||
| 1 | Ph | 57 | 42 | 93 | 39 | 90 |
| 2 | p-ClC6H4 | 55 | 43 | 95 | 40 | 94 |
| 3 | p-BrC6H4 | 55 | 46 | 93 | 40 | 97 |
| 4 | p-MeC6H4 | 50 | 49 | 88 | 46 | 92 |
| 5 | m-MeC6H4 | 57 | 41 | 93 | 42 | 96 |
下载: 导出CSV
表 5 吲哚与DA环丙烷的分子内立体选择性[3+2]环化反应[60]
Table 5. Stereoselective intramolecular [3+2] annulation reactions of DA cyclopropanes with indoles[60]
|
|
||||
| Entry | R1 | R2 | Yield/% | dr(49/50) |
| 1 | H | Et | 83 | 83/17 |
| 2 | H | Me | 87 | 74/26 |
| 3 | H | nHex | 77 | 78/22 |
| 4 | H | iPr | 77 | 90/10 |
| 5 | H | cHex | 83 | 88/12 |
| 6 | H | 3-Pent | 45 | 89/11 |
| 7 | H | tBu | trace | - |
| 8 | H | 1-Ad | trace | - |
| 9 | H | CH2tBu | 64 | 15/85 |
| 10 | H | CH2-1-Ad | 52 | 9/91 |
| 11 | H | 2-Ad | 61 | 6/94 |
| 12 | CH3 | iPr | 85 | 84/16 |
| 13 | CH3 | 2-Ad | 79 | 5/95 |
下载: 导出CSV
表 6 芳香氮杂亚胺叶立德与DA环丙烷的不对称[3+3]环加成反应的底物范围[74]
Table 6. Scope of the asymmetric [3+3] cycloadditions of aromatic azomethine imines with DA cyclopropane[74]
|
|
|||||
| Entry | 59 | Time/h | Yield/% | dr(cis/trans) | ee/% |
| 1 | 59a | 7 | 91 | >95/5 | 94 |
| 2 | 59b | 9 | 94 | >95/5 | 96 |
| 3 | 59c | 9 | 85 | 92/8 | 86 |
| 4 | 59d | 9 | 81 | 75/25 | 88/86 |
| 5 | 59e | 5.5 | 92 | 42/58 | 90/95 |
| 6 | 59f | 14 | 99 | 58/42 | 88/91 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们