Citation:
Yin Zhi-Jian, Wu Zong-Quan, Lin Feng, Qi Qiao-Yan, Xu Xiao-Na, Zhao Xin. A supramolecular bottlebrush polymer assembled on the basis of cucurbit[8]uril-encapsulation-enhanced donor-acceptor interaction[J]. Chinese Chemical Letters,
2017, 28(6): 1167-1171.
doi:
10.1016/j.cclet.2017.03.029

A supramolecular bottlebrush polymer assembled on the basis of cucurbit[8]uril-encapsulation-enhanced donor-acceptor interaction
English
A supramolecular bottlebrush polymer assembled on the basis of cucurbit[8]uril-encapsulation-enhanced donor-acceptor interaction
Abstract:
A supramolecular bottlebrush polymer has been constructed in water through the self-assembly of a rigid electron-deficient building block and an electron-rich monomer which bears two tetraethylene glycol chains, driven by CB[8]-encapsulation-enhanced donor-acceptor interaction. The as-formed supramolecular bottlebrush polymer has been characterized by 1H NMR titration experiment, UV-vis spectroscopy, DLS and 2D 1H NMR DOSY.
-
1. Introduction
Supramolecular polymer originates from the merging of supramolecular chemistry and polymer science [1]. Different from the classic polymers in which building parts are linked by covalent bonds, in supramolecular polymers, their components are connected through weak intermolecular interactions such as hydrogen-bonding [2-4], coordination interaction [5], host-guest interaction [6-9], donor-acceptor interaction [10, 11], C—H…π interaction [12], and dimerization of radicals [13, 14]. Since the first supramolecular polymer was reported by Lehn in 1990 [15], over the past decades a myriad of supramolecular polymers have been fabricated. Among them some interesting properties have been demonstrated and useful applications have been exploited. However, while supramolecular polymers play more and more important roles in the fabrication of functional soft materials, especially the so-call "smart materials" which can respond to the external stimuli [16, 17], their structural diversities are still quite limited in comparison with the traditional polymers.
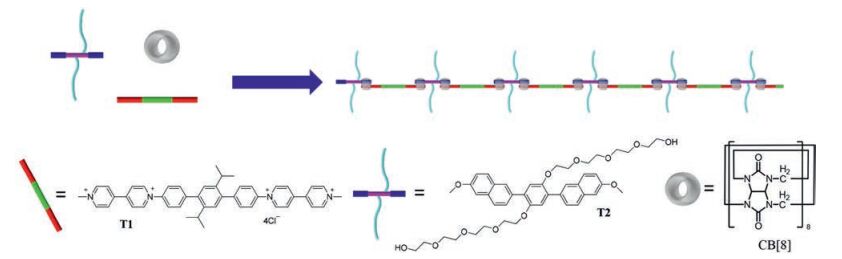
Polymer bottlebrushes, also known as molecular bottlebrushes, are a unique type of polymers which have drawn considerable attention in recent years [18]. A polymer bottlebrush consists of a long liner backbone on which short densely grafted side chains are attached. While traditional polymer bottlebrushes can be fabricated through grafting strategy [19], more recently self-assembly has emerged as a powerful tool to construct this unique type of polymeric materials, which lead to a new type of molecular bottlebrushes, that is, supramolecular bottlebrush polymers. In principle there are two approaches that can be applied to the construction of supramolecular bottlebrush polymers. The first one is to noncovalently attach side chains onto linear covalent backbones [20]. For the second approach, polymeric backbones are constructed through self-assembly driven by noncovalent bonds, while side chains are covalently linked to the building blocks of the backbones [21]. It is generally believed that the second approach is more useful because it is synthetically more accessible and easier to be implemented. However, so far most studies in this field are focused on utilizing hydrogen bonding [22] and aromatic stacking [23] as the main driven forces for the assembly of polymeric chains. To our surprise, cucurbit[8]uril(CB[8])-based host-guest interaction [24], as one of the most widely used driven forces for the fabrication of main-chain supramolecular polymers [25], has rarely been employed to construct supramolecular bottlebrush polymers. In this letter, we report the construction of a supramolecular bottlebrush polymer in water via CB[8]-encapsulation-enhanced donor-acceptor interaction between a rigid electron-deficient building block and an electron-rich monomer which bears two tetraethylene glycol chains. To the best of our knowledge, it is the first example of supramolecular bottlebrush polymer constructed through the CB[8]-based host-guest chemistry (Scheme 1).
Scheme1
2. Results and discussion
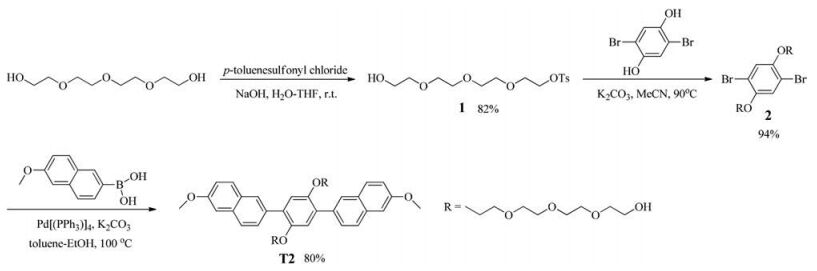
Compound T1 was prepared according to our previous work [26]. For the synthesis of compound T2, as shown in Scheme 2, the reaction of tetraethylene glycol and p-toluenesulfonyl chloride led to the formation of monosulfonate 1 in 82% yield, which was further heated with 2, 5-dibromohydroquinone in the presence of K2CO3 in anhydrous acetonitrile to afforded compound 2 in 94% yield. Compound 2 was then coupled with 6-methoxy-2-naphthaleneboronic acid to generate the target compound T2 in 80% yield.
Scheme2
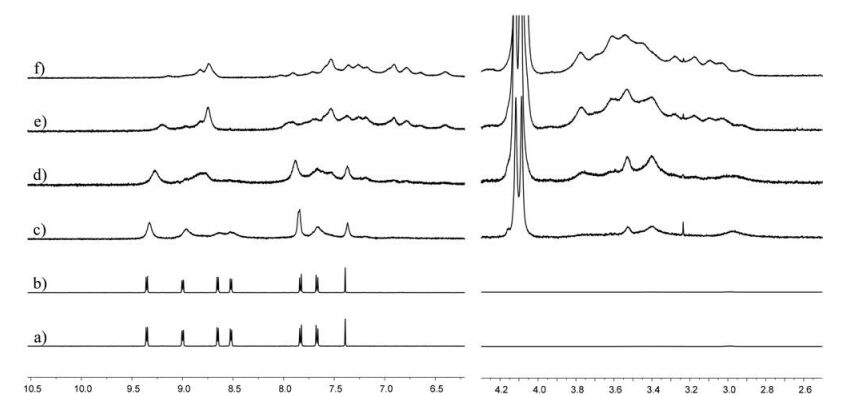
The self-assembly process was monitored by 1H NMR titration experiment. As can be seen in Fig. 1, compound T1 exhibits a highresolution spectrum, suggesting no self-aggregation of T1 itself in water. The addition of 1 equiv. of compound T2 results in almost no change of the spectrum of T1 and peaks corresponding to T2 can not be observed. It could be attributed to the extremely low solubility of T2 in water. However, upon the continuous addition of CB[8] into the mixture of T1 and T2, the peaks of T1 become broad and some new peaks appear. The peaks appear in the range of 7.50-6.25 ppm are attributed to the protons of naphthalene units of T2. However, they can not be unambiguously assigned due to the low resolution of the spectra. Moreover, the peaks corresponding to the tetraethylene glycol chains of T2 can also be observed in the range of 3.9-2.7 ppm, the intensities of which increase with the incremental addition of CB[8]. When 2 equiv. of CB[8] was added, the peaks of free T1 almost disappear and a spectrum with very broad peaks and low-resolution was obtained, indicating the formation of polymeric structures and a 1:1:2 stoichiometry for T1, T2 and CB[8].
图 1
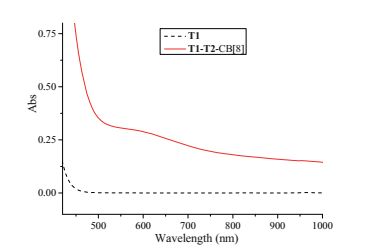
The formation of the supramolecular bottlebrush polymer is believed to be driven by donor-acceptor interaction between the electron-rich naphthalene unit and the electron-deficient viologen segment, which has already been well-established in the previous studies [27]. To acquire direct evidence for the existence of donor-acceptor interaction, UV-vis spectroscopy was carried out for the solution of a mixture of T1, T2 and CB[8] (1:1:2) in water. For comparison, UV-vis spectrum of compound T1 in water was also recorded. As shown in Fig. 2, while T1 exhibits almost no absorption in the region of 450-900 nm, the solution of T1, T2 and CB[8] (1:1:2) displays a broad absorption peak between 500-700 nm. It is the typical charge-transfer absorption resulted from donor-acceptor interaction [27], confirming the driven force for the formation of the supramolecular bottlebrush polymer is donor-acceptor interaction between the naphthalene units of T2 and the viologen segments of T1.
图 2

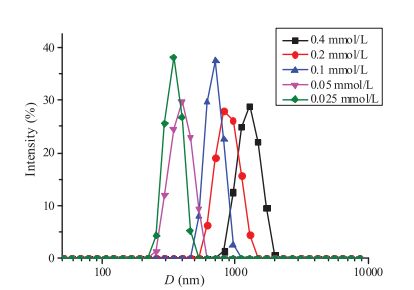
The formation of the supramolecular bottlebrush polymer in water has been further evidenced by the dynamic light scattering (DLS) experiment (Fig. 3). Increasing the concentration of the mixture of T1, T2 and CB[8] (1:1:2) leads to increase of the hydrodynamic diameters of the aggregates formed in solution, indicating a higher degree of polymerization at higher concentration. For example, the aggregates in water have an average hydrodynamic radius of around 340 nm at the concentration of T1 being 0.025 mmol/L. The value increases to ca. 710 nm at a T1 concentration of 0.1 mmol/L. When the concentration of T1 increases to 0.4 mmol/L, the aggregates with an average hydrodynamic radius of around 1300 nm were observed.
图 3

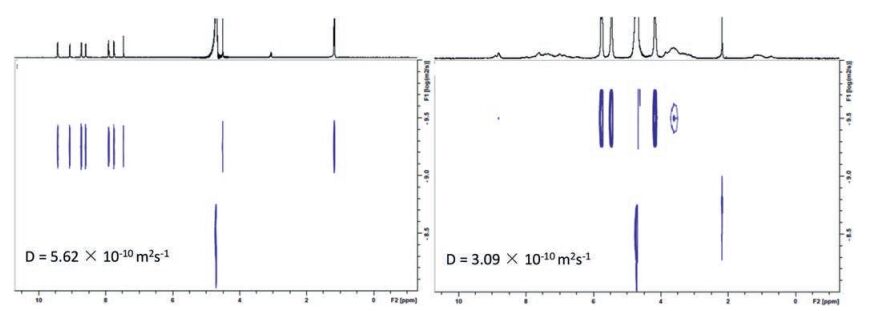
2D 1H NMR diffusion ordered spectroscopy (DOSY) was also carried out to characterize the polymeric structures formed in water. It was found that T1 itself has a value of diffusion coefficient (D) to be 5.62 ×10-10 m2/s at a concentration of 1.0 mmol/L in D2O. However, the solution of the mixture of T1, T2 and CB[8] (1:1:2) in D2O displays a diffusion coefficient of 3.09 ×10-10 m2/s at the same concentration, strongly suggesting the formation of the expected supramolecular bottlebrush polymer (Fig. 4).
图 4

3. Conclusion
In summary, we have successfully constructed a supramolecular bottlebrush polymer in water by taking advantage of CB[8]-encapsulation-enhanced donor-acceptor interaction between naphthalene units and the viologen segments. The as-prepared supramolecular bottlebrush polymer has a rigid supramolecular polymeric backbone as the handle and flexible tetraethylene glycols as the bristles. Compared with its analogs reported in literature which are fabricated in organic solvents, supramolecular bottlebrush polymers assembled in water is more attractive because they are more biocompatible and thus have potential applications in the field of biomaterials. On the other hand, construction of bottlebrush polymers through CB[8]-based selfassembly is facile and the resulting polymeric structures are extremely stable. In this context, fabricating more supramolecular bottlebrush polymers in aqueous phase and further exploiting their applications are expected in the future.
4. Experimental
Synthesis of compound 1: NaOH (2.74 g, 68 mmol) in water (15 mL) was added to a THF (15 mL) solution of tetraethylene glycol (60 g, 309 mmol). p-Toluenesulfonyl chloride (8.4 g, 44 mmol) in 50 mL of THF was added slowly when the reaction mixture was kept in an ice bath. After stirring for 5 h, the reaction mixture was poured into 250 mL of ice water. The organic layer was separated, and the aqueous layer was extracted with dichloromethane. The combined organic layer was washed with water and dried over anhydrous MgSO4, and the solid was filtered. The solvent was evaporated under reduced pressure to afford 1 as a clear oil (12.6 g, 82%). 1H NMR (500 MHz, CDCl3): δ 7.80 (d, 2H, J = 8.1 Hz), 7.34 (d, 2H, J = 8.1 Hz), 4.16 (t, 2H, J = 4.9 Hz), 3.59-3.74 (m, 14H), 2.45 (s, 3H).
Synthesis of compound 2: A mixture of 2, 5-dibromohydroquinone (500 mg, 1.87 mmol), K2CO3 (1.03 g, 7.45 mmol) and anhydrous acetonitrile (20 mL) was refluxed under an argon atmosphere for 1 h and then a solution of 1 (1.62 g, 4.65 mmol) in anhydrous acetonitrile (5 mL) was added dropwise within 0.5 h. After completing the addition, the reaction mixture was stirred for 24 h. The solvent was removed and the residue was treated with 10% aqueous HCl and then extracted with dichloromethane. The organic layer was washed with water three times and dried over anhydrous MgSO4. After filtration and evaporation, the crude was purified by flash column chromatography (dichloromethane/ methanol 200:3) to give compound 2 (1.15 g, 94%) as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.16 (s, 2H), 4.13-3.60 (m, 32H).
Synthesis of compound T2: To a mixture of 6-methoxy-2-naphthaleneboronic acid (147 mg, 0.73 mmol), compound 2 (150 mg, 0.24 mmol), and Pd[P(Ph3)]4 (28 mg, 0.024 mmol) in a 25 mL flask, a mixture of toluene (3 mL), ethanol (1 mL) and 2 mol/L potassium carbonate aqueous solution (0.2 mL) was added. The mixture was degassed through three freeze-pump-thaw cycles under an argon atmosphere and then stirred at 100 ℃ for 48 h. After being cooled to room temperature, the reaction mixture was extracted with dichloromethane. The organic layer was washed with water, dried over MgSO4, filtered and evaporated. The crude product was purified by flash column chromatograph (dichloromethane/methanol 40:1) to give compound T2 (150 mg, 80%) as a gray solid. 1H NMR (500 MHz, CDCl3): δ 8.00 (s, 2H), 7.79 (d, 2H, J = 9.7 Hz), 7.77 (d, 4H, J = 0.9 Hz), 7.20-7.16 (m, 4H), 7.15 (s, 2H), 4.16-4.10 (m, 4H), 3.96 (s, 6H), 3.78-3.71 (m, 4H), 3.69-3.63 (m, 4H), 3.62-3.48 (m, 20H). 13C NMR (125 MHz, CDCl3): δ 157.77, 150.65, 133.62, 133.58, 131.06, 129.67, 128.89, 128.52, 128.00, 126.11, 118.80, 117.12, 105.59, 72.38, 70.78, 70.56, 70.29, 69.85, 69.56, 61.72, 55.36. MS (ESI): m/z 775.6 [M]+. HRMS (ESI): Calcd. for C44H54O12 [M]+: 775.3688. Found: 775.3685.
5. Acknowledgment
We thank the National Natural Science Foundation of China (No. 21402228) for financial support.
-
-
[1]
Yang L., Tan X., Wang Z., Zhang X.. Supramolecular polymers:historical development[J]. preparation, characterization, and functions, Chem. Rev., 2015, 115: 7196-7239.
-
[2]
Xiao T., Feng X., Ye S.. Highly controllable ring-chain equilibrium in quadruply hydrogen bonded supramolecular polymers[J]. Macromolecules, 2012, 45: 9585-9594. doi: 10.1021/ma302459n
-
[3]
Du P., Kong J., Wang G.. Hydrogen bonded supramolecular polymers in both apolar and aqueous media:self-assembly and reversible conversion of vesicles and gels[J]. Chin. J. Chem., 2011, 29: 2597-2605. doi: 10.1002/cjoc.201100254
-
[4]
Zhan T.G., Zhou T.Y., Qi Q.Y.. The construction of supramolecular polymers through anion bridging:from frustrated hydrogen-bonding networks to well-ordered linear arrays[J]. Polym. Chem., 2015, 6: 7586-7593. doi: 10.1039/C5PY01284H
-
[5]
Tian D., Liu X.J., Chen R.Y., Zhang Y.H.. Syntheses. structures, luminescent and magnetic properties of two coordination polymers based on a flexible multidentate carboxylate ligand[J]. Chin. Chem. Lett., 2015, 26: 499-503. doi: 10.1016/j.cclet.2015.01.019
-
[6]
Su Y.S., Liu J.W., Jiang Y., Chen C.F.. Assembly of a self-complementary monomer:formation of supramolecular polymer networks and responsive gels[J]. Chem. Eur. J., 2011, 17: 2435-2441. doi: 10.1002/chem.201002862
-
[7]
Dong S., Zheng B., Wang F., Huang F.. Supramolecular polymers constructed from macrocycle-based host-guest molecular recognition motifs[J]. Acc. Chem. Res., 2014, 47: 1982-1994. doi: 10.1021/ar5000456
-
[8]
Qian H., Guo D.S., Liu Y.. Cucurbituril-modulated supramolecular assemblies:from cyclic oligomers to linear polymers[J]. Chem. Eur. J., 2012, 18: 5087-5095. doi: 10.1002/chem.v18.16
-
[9]
Cao T.T., Yao X.Y., Zhang J., Wang Q.C., Ma X.. A cucurbit[8] uril recognized rigid supramolecular polymer with photo-stimulated responsiveness[J]. Chin. Chem. Lett., 2015, 26: 867-871. doi: 10.1016/j.cclet.2015.01.032
-
[10]
Burattini S., Greenland B.W., Hayes W.. A supramolecular polymer based on tweezer-type π-π stacking interactions:molecular design for healability and enhanced toughness[J]. Chem. Mater., 2011, 23: 6-8. doi: 10.1021/cm102963k
-
[11]
Tian Y.K., Shi Y.G., Yang Z.S., Wang F.. Responsive supramolecular polymers based on the bis[alkynylplatinum(Ⅱ)] terpyridine molecular tweezer/arene recognition motif[J]. Angew. Chem. Int. Ed., 2014, 53: 6090-6094. doi: 10.1002/anie.201402192
-
[12]
Zhang Z., Luo Y., Chen J.. Formation of linear supramolecular polymers that is driven by C-H…π interactions in solution and in the solid state[J]. Angew. Chem. Int. Ed., 2011, 50: 1397-1401. doi: 10.1002/anie.v50.6
-
[13]
Chen L., Zhang Y.C., Wang W.K.. Conjugated radical cation dimerizationdriven generation of supramolecular architectures[J]. Chin. Chem. Lett., 2015, 26: 811-816. doi: 10.1016/j.cclet.2015.01.036
-
[14]
Zhan T.G., Zhou T.Y., Lin F.. Supramolecular radical polymers selfassembled from the stacking of radical cations of rod-like viologen di-and trimers[J]. Org. Chem. Front., 2016, 3: 1635-1645. doi: 10.1039/C6QO00298F
-
[15]
Fouquey C., Lehn J.M., Levelut A.M.. Molecular recognition directed selfassembly of supramolecular liquid crystalline polymers from complementary chiral components[J]. Adv. Mater., 1990, 2: 254-257. doi: 10.1002/(ISSN)1521-4095
-
[16]
Xu J.F., Chen Y.Z., Wu D.. Photoresponsive hydrogen-bonded supramolecular polymers based on a stiff stilbene unit[J]. Angew. Chem. Int. Ed., 2013, 52: 9738-9742. doi: 10.1002/anie.201303496
-
[17]
Qu D.H., Wang Q.C., Zhang Q.W., Ma X., Tian H.. Photoresponsive host-guest functional systems[J]. Chem. Rev., 2015, 115: 7543-7588. doi: 10.1021/cr5006342
-
[18]
Zhang M., A.Müller H.E.. Cylindrical polymer brushes[J]. J. Polym. Sci. A:Polym. Chem., 2005, 43: 3461-3481. doi: 10.1002/(ISSN)1099-0518
-
[19]
Sheiko S.S., Sumerlin B.S., Matyjaszewski K.. Cyclindrical molecular brushes:synthesis. characterization, and properties[J]. Prog. Polym. Sci., 2008, 33: 759-785. doi: 10.1016/j.progpolymsci.2008.05.001
-
[20]
Liu S., Chen Q., Sheng Y.. Unraveling the forming mechanism of hierarchical helices via self-assembly of an achiral supramolecular polymer brush[J]. Polym. Chem., 2015, 6: 3926-3933. doi: 10.1039/C5PY00163C
-
[21]
Catrouillet S., Bouteiller L., Boyron O.. Patchy supramolecular bottlebrushes formed by solution self-assembly of bis(urea)s and tris(urea)s decorated by two incompatible polymer arms[J]. Langmuir, 2016, 32: 8900-8908. doi: 10.1021/acs.langmuir.6b01609
-
[22]
Catrouillet S., Brendel J.C., Larnaudie S.. Tunable length of cyclic peptidepolymer conjugate self-assemblies in water[J]. ACS Macro Lett., 2016, 5: 1119-1123. doi: 10.1021/acsmacrolett.6b00586
-
[23]
Dingenouts N., Klyatskaya S., Rosenfeldt S., Ballauff M., Höger S.. Temperatureinduced switching between aggregated and nonaggregated states in coil-ringcoil block copolymers[J]. Macromolecules, 2009, 42: 5900-5902. doi: 10.1021/ma901022w
-
[24]
Lagona J., Mukhopadhyay P., Chakrabarti S., Isaacs L.. The cucurbit[n]uril family[J]. Angew. Chem. Int. Ed., 2005, 44: 4844-4870. doi: 10.1002/(ISSN)1521-3773
-
[25]
Liu Y., Yang H., Wang Z., Zhang X.. Cucurbit [
8 ] uril-based supramolecular polymers[J]. Chem. Asian J., 2013, 8: 1626-1632. doi: 10.1002/asia.v8.8 -
[26]
Fan Y., Lin F., Xu X.N., Xu J.Q., Zhao X.. Construction of a rod-coilsupramolecular copolymer through CB [
8 ] -encapsulation-enhanced donor-acceptor interaction[J]. Acta Polym. Sin, 2017, : 80-85. -
[27]
Kim H.J., Heo J., Jeon W.S.. Selective inclusion of a hetero-guest pair in a molecular host:formation of stable charge-transfer complexes in cucurbit [
8 ] uril[J]. Angew. Chem. Int. Ed., 2001, 40: 1526-1529. doi: 10.1002/(ISSN)1521-3773
-
[1]
-


Figure 2 UV-vis spectra of T1 and T1-T2-CB[8] (1:1:2) in water at 25 ℃. The concentration of T1 was 0.2 mmol/L.
Figure 3 DLS profile of T1-T2-CB[8] (1:1:2) in water at different concentrations of T1.
-

 下载:
下载:

 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 2251
- HTML全文浏览量: 86
