College of Chemical Engineering, Fuzhou University, Fuzhou 350116, China
b.
State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China
c.
Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou 350108, China
Received Date:
16 September 2021 Accepted Date:
01 November 2021 Revised Date:
21 October 2021 Available Online:
15 June 2022
Abstract:
Aggregation-induced emission (AIE) based luminescent materials are generating intensive interest due to their unique fluorescence in the aggregation state. Herein we report a strategy of dynamic covalent chemistry (DCC) controlled AIE luminogens for the regulation of multicolor emission in reversible covalent polymer networks. Tetraphenylethene derived ring-chain tautomers were prepared, and the emission was readily controlled through multimode, such as changing the solvent, adding the base, and dynamic covalent reactions with amines. Moreover, the construction of dynamic covalent cross-linked luminescent hydrogels with tunable fluorescent, self-healing, and mechanical properties, was realized. The combination of AIE and aggregation-caused quenching (ACQ) fluorophores in the polymer network further enabled the realization of a multicolor modulator, including white emission, in both solution and gel states. The strategies and results presented should find utility in dynamic assemblies, polymer networks, chemical sensing, and responsive materials.
Luminescent materials have received extensive attention and found application in chemical sensing, biological imaging, information storage, and organic light-emitting diodes [1-8]. Multicolor emissive systems with tunable wavelength, especially those tuned for white light emission, are of significance due to their essential role in lighting and displays [9-16]. Therefore, the development of novel strategies for the creation of multicolor systems with wide-spectrum emission, outstanding switchablility, as well as multistimuli responsiveness is highly desirable. Recently supramolecular self-assemblies driven by noncovalent interactions, such as host-guest complexes and metal coordination, have been constructed for the control of color-conversion processes [17-23]. In contrast to conventional fluorophores usually suffering from aggregation-caused quenching (ACQ) effect, fluorescent materials with aggregation-induced emission (AIE) [24-31] attributes are of notable interest owing to their high fluorescence quantum yields in the aggregated form, greatly promoting their utility in solid state [32-36].
Dynamic covalent chemistry (DCC) [37-42], allowing the reversible combination of molecular building blocks via the breakage and reformation of covalent bonds, has become a powerful tool for the construction of functional systems, including dynamic assemblies, ordered frameworks, and complex architectures [43−48]. In particular, the introduction of reversible covalent bonds into polymers can lead to the creation of dynamic covalent polymer networks, endowing them with unique properties, such as adaptability, self-healing, and shape-memory [49−55]. Recently, a variety of fluorogens were incorporated into dynamic covalent systems for regulating fluorescent signaling outputs [56−61]. For example, temperature-responsive dynamic polymer networks were reported, and their emission was regulated by reversible Diels-Alder reactions [62−65]. In addition, by introducing aldehyde derived tetraphenylethene (TPE) units to molecular scaffolds through imine or hydrazone linkages, a suit of dynamic covalent assemblies showing AIE effect were attained [66−71]. As a result, DCC would offer ample opportunities for controlling emission conversion processes.
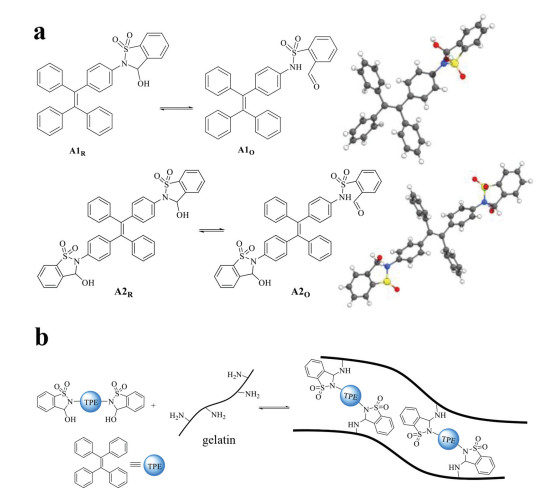
In the present work, we designed a type of TPE derived stimuli-responsive AIE luminogens (Fig. 1a) through the strategy of ring-chain isomerization, which plays a critical role in harnessing fluorophores of fluorescein and rhodamine [72, 73], as well as photoswitches of diarylethene [74, 75] and spiropyran [76, 77]. In conjunction to the manipulation of TPE with solvent effect, ring-chain tautomerism of aldehyde and its intermolecular dynamic covalent reactions (DCRs) with amines provided additional handles for control. Moreover, the combination of bifunctional aldehyde with polyamines enabled the formation of cross-linked luminescent hydrogels (Fig. 1b). Finally, dynamic polymer networks were constructed to achieve addressable multicolor systems with wide-spectrum emission, including white color, and vapor-induced luminescent switching was further realized.
Figure 1
Figure 1.
(a) Structures of A1, A2 and their ring-chain tautomerization equilibrium, with crystal structures listed. (b) The construction of luminescent dynamic covalent polymernetworks. Note: "A" is the abbreviation of aldehyde, and the subscripts "R" and "O" represent the ring form and open form, respectively.
Building upon our strategy of dual reactivity based DCC [78−81], TPE luminogens were attached with 2-formylbenzenesulfonamide to afford a ring-chain tautomerization equilibrium between aldehyde and its cyclic hemiaminal. Monoaldehyde A1 and dialdehyde A2 (Fig. 1a) were prepared from their corresponding amines and o-formylbenzenesulfonyl chloride (Schemes S1 and S2 in Supporting information) and fully characterized (Figs. S1−S4 in Supporting information). X-ray crystal analysis confirmed cyclic hemiaminal structures for A1 and A2 (Fig. 1a). A dihedral angle of −86° was apparent between the phenyl ring of TPE and sulfonamide nitrogen of A1, suggesting the lack of p-π conjugation.
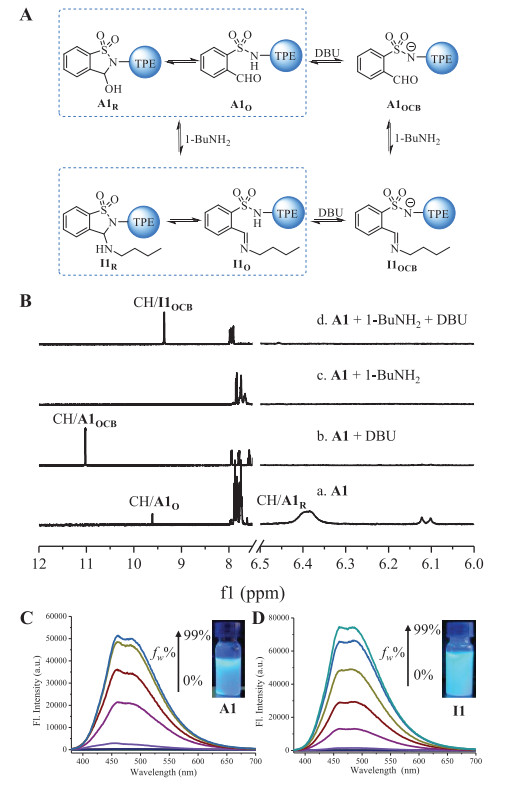
The intramolecular ring-chain equilibrium of A1 and A2 was also examined in solution (Fig. 2A). 1H NMR spectra revealed that the cyclic tautomer accounts for the major population of both A1 and A2 in CD3CN (Figs. S1−S4). Taking A1 as an example (Fig. 2B-a), the ring form (A1R, 90%) was detected with the hemiaminal methine proton at 6.4 ppm, along with a small amount (10%) of aldehyde (A1O, CHO around 9.6 ppm). In order to obtain the open aldehyde form, base DBU was titrated into A1 to shift ring-chain equilibrium (Fig. 2A). Upon the addition of DBU (2.0 equiv.) the appearance of the formyl proton around 11.0 ppm in 1H NMR spectrum indicated the formation of the open conjugate base of A1 (A1OCB, Fig. 2B-b). A1 was recovered through the addition of acid to A1OCB (Fig. S6 in Supporting information).
Figure 2
Figure 2.
(A) The conversion of A1 triggered by DBU and 1-BuNH2. (B) 1H NMR spectra of A1 in (a) CD3CN, (b) with the addition of DBU (2.0 equiv.), (c) its reaction with 1-BuNH2 (3.0 equiv.), and (d) the addition of DBU (2.0 equiv.) into (c). (C) Fluorescencespectra of A1 (50 µmol/L) in ACN/H2O. (D) Fluorescence spectra of I1 (50 μmol/L) in ACN/H2O. Inset: photograph under a 365 nm UV lamp. λex = 340 nm. Note: "I" is the abbreviation of imine.
Attention was then turned to dynamic covalent reactions with amines. The reaction of A1 with 1-butylamine (3.0 equiv.) proceeded rapidly within 3 min in the presence of molecular sieves (MS) in CD3CN, affording cyclic aminal I1 in quantitative yield (Fig. 2B-c and Fig. S10 in Supporting information). The fast reaction is likely due to intramolecular acid catalysis caused by the neighboring acidic sulfonamide group. It is notable that the resonance of aminal methine CH of I1 overlapped with aromatic peaks. Gratifyingly, anionic open imine form I1OCB emerged with DBU (2.0 equiv.) present, as evidenced by the methine CH at 9.3 ppm(Fig. 2B-d). Similar reactivity with amine and associated ring-chain tautomers were found with A2 (Figs. S9 and S11 in Supporting information).
We next set to explore optical properties of aldehydes and their corresponding aminals/imines. As depicted in Fig. 2C, fluorescence intensity at 460 nm was enhanced while water was titrated into a solution of A1 in acetonitrile (ACN), with an increasement of around 800 fold at water fraction (fw) of 99% in ACN/water and showing typical AIE characteristics. The strong blue emission under a UV lamp validated AIE property of A1. DBU was then added to a solution of A1 in a mixed solution of ACN/water (fw = 80%). The blue emission at 460 nm was gradually quenched with increasing amount of DBU (Fig. S14 in Supporting information). In situ created I1 from A1 and 1-butylamine was also subjected for fluorescence studies. Analogous AIE feature was revealed, with emission maximum at 460 nm (Fig. 2D). Moreover, DBU induced quenching took place for I1 (Fig. S15 in Supporting information). Base and amine triggered fluorescence change was further verified by significantly different quantum yields (ϕ) of solutions of A1 (21.9%), A1OCB(< 0.1%), I1 (16.7%) and I1OCB (< 0.1%) (Table S2 in Supporting information). Fluorescence modulation was also feasible for A2 and its imine derivative I2 (Figs. S16-S19 in Supporting information). In addition, reversible fluorescence switching was achieved with several cycles by using base/acid (Figs. S20 and S21 in Supporting information).
Although these results fall in line with AIE behaviors of TPE unit, the fluorescence can be readily regulated by the reaction with amines and the variation of pH. Base induced quenching was explained as following: while the base converts the cyclic form into the conjugate base of open aldehyde/imine, the aggregation of fluorophore was prevented by the negative charge in conjugate base due to electrostatic repulsion. In addition, the electron density of TPE fluorophore is pulled away by electron-withdrawing 2-formylbenzenesulfonyl group, contributing to the quenching of the original emission [80].
By utilizing the AIE feature of TPE derived luminogens, solid-state fluorescence was next measured. Both powder A1 and A2 exhibited strong blue emission around 460 nm (Figs. S22 and S23 in Supporting information). A lower quantum yield for A2 (1.95%) than A1 (3.91%) was in accordance with the data in solution and X-ray structures. The dihedral angle between one unsubstituted phenyl group and the C=C bond of TPE in A2 was −97°, while A1 has a much smaller dihedral angle (−55°, Fig. S5 in Supporting information). The varying conformation of A1 and A2 would lead to the difference in the emission: the increase of dihedral angle would reduce the quantum yield due to poor conjugation in the molecule [82−84].
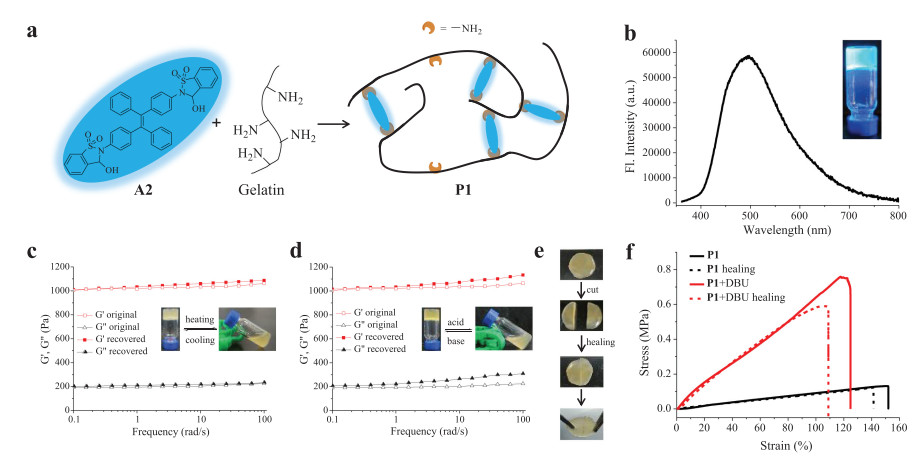
Having examined luminescent properties of aldehydes/imines, dynamic covalent polymer network was constructed with dialdehyde A2 as a crosslinking agent for a polyamine (Fig. 3a). Responsive luminescent gels have emerged as promising intelligent materials by virtue of their sensitive signal change in response to external stimuli, especially for those gels with good mechanical toughness and self-healing ability [85, 86]. Toward this end, a hydrogel P1 was prepared from A2 and gelatin, which is derived from collagen with low cost and shows high film-forming ability as well as biocompatibility and degradability [87, 88]. Gratifyingly, P1 exhibited strong blue-green emission at 500 nm (Fig. 3b).
Figure 3
Figure 3.
(a) The illustration of a dynamic covalent polymernetwork created by A2 and gelatin. (b) Fluorescencespectra of gel P1 (λex = 360 nm) and its photograph under a 365 nm UV lamp. Frequency sweep measurement of original and recovered gel P1 triggered by (c) temperature or (d) pH change. Definitions: storagemodulus, G′; lossmodulus, G′′. Inset: photographs of phase transition. (e) Photographs of self-healing process of P1. (f) Stress-strain curves of P1 and its regulation by DBU. Note: "P" is the abbreviation of polymergel.
Sol-gel transitions were next studied. The storage modulus (G′) of gel P1 was higher than the loss modulus (G″) as measured by the frequency sweep in rheology test at 25 ℃, which further validated the formation of solid-like polymer (Fig. 3c). The gel state of P1 readily converted to the sol state (G′ < G″) upon heating, with an apparent crossover point of G′ and G″ at 40 ℃ (Fig. S25 in Supporting information). The gel state was reformed while cooled to room temperature (Fig. 3c). The sol-gel transition was also achieved by changing pH (Fig. 3d). Upon the addition of HCl gel collapse was observed due to the breakage of imine bonds, as confirmed by the emergence of aldehyde peak in 1H NMR spectrum (Fig. S27 in Supporting information). Imine bonds were recovered to recreate the gel state by increasing pH using DBU, which was supported by the maintenance of G′ and G″ in rheology analysis (Fig. 3d). Such a pH or temperature-induced phase transition could be realized several cycles through consecutive addition of acid/base or during a heating/cooling process (Fig. S26 in Supporting information), suggesting great reversibility of the sol-gel transition.
The healability was then investigated by taking use of the dynamic nature of imine bond. A freshly prepared gel was cut into two pieces, and these pieces were then put together for keeping in close contact. The reformation of complete gel was observed after 5 min without external force, indicative of excellent self-healing capability (Fig. 3e). Tensile experiments were further conducted to quantify mechanical properties. As shown in Fig. 3f, an intact gel P1 was stretched up to 2.5 times of its original length. The healing efficiency was found to be around 90% based on tensile test. A comparison of stress-strain curves of P1 and P1-DBU gave interesting results. While DBU converted cyclic hemiaminal to open imine in polymer network, a strikingly different stress-strain relationship with increased fracture stress and reduced stretchability was found. The enhanced strength of imine double bonds within crosslinks could contribute to varying mechanical features between P1 and P1-DBU. As expected, the emission of gel was quenched with DBU (Fig. S28 in Supporting information). These findings demonstrate that both luminescent and mechanical properties can be facilely tuned.
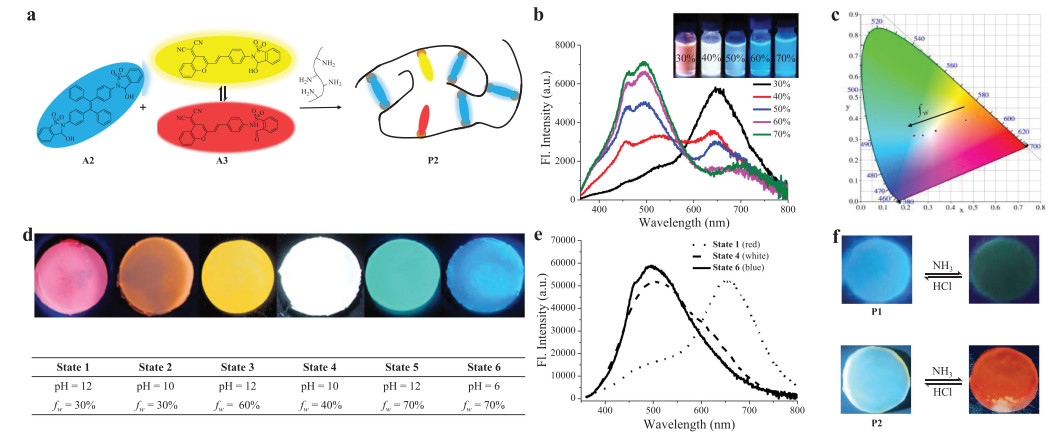
The next goal was to realize a multicolor emissive system, including white emission. Toward this end, a co-assembly strategy was developed to broaden the spectral range. An ACQ-type fluorescent switch A3 [80], which was reported by us recently and is able to generate yellow (560 nm) and red emission (709 nm) in the closed and open form, respectively (Fig. S29 in Supporting information), was utilized to co-assemble into a dynamic covalent polymer network of dialdehyde A2 and gelatin (Fig. 4a). In contrast to A1/I1, the addition of base into A3/3I resulted in an increase in red fluorescence (Fig. S30 in Supporting information) due to enhanced electron-withdrawing ability of dicyanomethylenebenzopyran based fluorophore. In light of different spectral range and emitting mechanism of A2/I2 and A3/I3 as well as the sensitivity of imine bonds to acid, the fluorescence of imine polymer network from A2, A3, and gelatin could be controlled by manipulating the aggregation state of luminogens through altering water fraction (fw) and by modulating the reversible covalent bonds through pH change, thereby leading to advanced dual regulation for multicolor emission (Fig. S31 in Supporting information).
Figure 4
Figure 4.
(a) The creation of a multicolor polymernetwork. (b) Fluorescent spectra of polymer P2 under varying water fraction in DMSO/water, with fluorescent color change shown in the inset (λex = 360 nm). (c) CIE coordinates of the multicolor system with varying fw on CIE 1931 chromaticity diagram. (d) Photographs of fluorescent color change of gel P2 with varying pH and water fraction. (e) Fluorescent spectra of gel P2 under different states (λex = 360 nm). (f) Photographs of fluorescent color change of gel P1 or P2 in response to acid/base vapors.
As a proof-of-concept, aldehydes A2, A3 and gelatin (10:1:200, w/w/w) were mixed in a solution of DMSO/H2O (f = 30%, pH 10), and dynamic covalent reactions of aldehydes with amine allowed the formation of imine polymer network P2, affording an orange emission with a CIE coordinate of (0.47, 0.39) (Figs. 4b and c). With the addition of water, blue emission of imines from A2 was activated due to AIE effect, while red emission of imines from A3 was suppressed because of ACQ nature (Fig. 4b). As a result, the color gradually shifted from orange to blue emission region, as plotted in CIE 1931 chromaticity diagram (Fig. 4c). Almost pure white light emission (0.33, 0.34) was obtained at fw of 40%.
Fluorescence switching of polymer networks was also achieved by changing pH (Figs. S32−S41 in Supporting information). For example, A2, A3, and gelatin (10:1:200, w/w/w) was mixed in DMSO/water (6:4) at pH 2. A3 was almost non-fluorescent at acidic pH (Fig. S33), and the blue emission of A2 was observed. By increasing pH the imine network between gelatin and aldehydes was formed, and the red emission was turned on by imines of A3. Therefore, the fluorescent color gradually changed from blue to white emission. By further enhancing pH to give conjugate bases of open imines the blue emission of TPE units was quenched while the red emission from imines of A3 was maintained.
On account of diverse luminescent behaviors in solution, gel P2 prepared from A2, A3 and gelatin was next studied to afford multicolor emitters. As indicated in Fig. 4d, fluorescent colors of the gel were regulated from red to blue through varying pH and f, consistent with the features observed in solution. It is noteworthy that almost pure white emission was obtained when the gel was formed at pH 10 and fw of 40%, and a CIE value of (0.31, 0.35) was obtained based on the emission spectra (Fig. 4e). Finally, to showcase the application potential of fluorescent gel the emission switching in response to vapors was conducted (Fig. 4f). For example, gel P1 was placed onto NH3 vapor, and the fluorescence was turned off after 5 min. The exposure to HCl vapor resulted in the recovery of the blue emission. Furthermore, the interconversion between blue and orange emission was feasible when P2 was in contact with HCl/NH3 vapors. The switchability was confirmed through multiple cycles of exposure to HCl/NH3 vapors (Fig. S42 in Supporting information).
In summary, a new type of stimuli-responsive dynamic covalent luminogens exhibiting AIE were developed based on ring-chain tautomerism design. The manipulation of open/cyclic isomers was realized through changing pH and dynamic covalent reactions with amines, allowing the modulation of emission. Moreover, polymeric hydrogels with tunable fluorescence and self-healing properties were prepared through dialdehyde A2 and gelatin. By taking advantage of unique mechanism of AIE and ACQ fluorophores, dynamic covalent polymer networks were further constructed to achieve tunable multicolor modulators, and white emission was attained in both solution and gel phases. Due to the significance of AIE materials, the results presented herein should afford opportunities for molecular assemblies, polymer networks, and intelligent materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank National Natural Science Foundation of China (NSFC, Nos. 21672214, 22071247 and 22101283), the Strategic Priority Research Program (No. XDB20000000) and the Key Research Program of Frontier Sciences (No. QYZDB-SSW-SLH030) of the CAS, NSF of Fujian Province (No. 2020J06035), and Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (No. 2021ZR112) for funding.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.001.
[1]
Y. Yang, Q. Zhao, W. Feng, F. Li, Chem. Rev.113 (2013) 192-270. doi: 10.1021/cr2004103
Figure 1
(a) Structures of A1, A2 and their ring-chain tautomerization equilibrium, with crystal structures listed. (b) The construction of luminescent dynamic covalent polymernetworks. Note: "A" is the abbreviation of aldehyde, and the subscripts "R" and "O" represent the ring form and open form, respectively.
Figure 2
(A) The conversion of A1 triggered by DBU and 1-BuNH2. (B) 1H NMR spectra of A1 in (a) CD3CN, (b) with the addition of DBU (2.0 equiv.), (c) its reaction with 1-BuNH2 (3.0 equiv.), and (d) the addition of DBU (2.0 equiv.) into (c). (C) Fluorescencespectra of A1 (50 µmol/L) in ACN/H2O. (D) Fluorescence spectra of I1 (50 μmol/L) in ACN/H2O. Inset: photograph under a 365 nm UV lamp. λex = 340 nm. Note: "I" is the abbreviation of imine.
Figure 3
(a) The illustration of a dynamic covalent polymernetwork created by A2 and gelatin. (b) Fluorescencespectra of gel P1 (λex = 360 nm) and its photograph under a 365 nm UV lamp. Frequency sweep measurement of original and recovered gel P1 triggered by (c) temperature or (d) pH change. Definitions: storagemodulus, G′; lossmodulus, G′′. Inset: photographs of phase transition. (e) Photographs of self-healing process of P1. (f) Stress-strain curves of P1 and its regulation by DBU. Note: "P" is the abbreviation of polymergel.
Figure 4
(a) The creation of a multicolor polymernetwork. (b) Fluorescent spectra of polymer P2 under varying water fraction in DMSO/water, with fluorescent color change shown in the inset (λex = 360 nm). (c) CIE coordinates of the multicolor system with varying fw on CIE 1931 chromaticity diagram. (d) Photographs of fluorescent color change of gel P2 with varying pH and water fraction. (e) Fluorescent spectra of gel P2 under different states (λex = 360 nm). (f) Photographs of fluorescent color change of gel P1 or P2 in response to acid/base vapors.

 DownLoad:
DownLoad:

 下载:
下载:





