

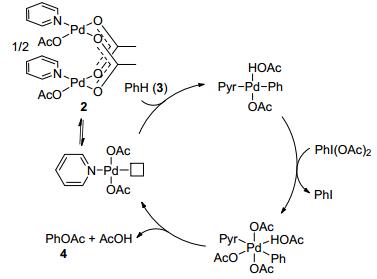
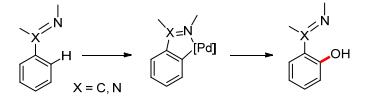
Scheme 1.
Proposed catalytic cycle of palladium-catalyzed PhI- (OAc)2 oxidized aromatic C—H acetoxylation
Transition Metal-Catalyzed Regio-selective Aromatic C—H Bond Oxidation for C—O Bond Formation
Fanzhi Yang , Han Zhang , Xuri Liu , Bo Wang , Lutz Ackermann

Phenols are important structural motifs in the research areas of pharmaceuticals, agrochemicals, polymers, and natural products. There has been a growing demand for developing versatile synthetic methodologies of phenol derivatives.[1] Recently, transition metal-catalyzed direct aromatic C—H oxidation has emerged as a powerful tool in the regio-selective synthesis of phenols. This strategy compares favorably to classical phenol synthesis with respect to the atom-economy as well as the step-economy.
There have already been several distinguished reviews on the research progress of transition metal-mediated aromatic C(sp2)—H oxygenation from different perspectives: Sanford et al. reviewed C—O bond formation as a section in the subject of palladium-catalyzed C—H functionalizations.[2] Later, Enthaler and Company summarized the progress in palladium-catalyzed hydroxylation and alkoxylation.[3] Ackermann et al. reviewed the recent advances in ruthenium-catalyzed direct hydroxylation of C(sp2)—H and C(sp3)—H bonds.[4] Baran et al. reviewed the oxidation of both C(sp2)—H and C(sp3)—H bonds, emphasizing the chemoselectivity imposed by the nature of the substrates, such as inductive effects, conjugation, hyper-conjugation, steric hindrance, and strain release.[5] Regarding the oxidants, Jiao et al. summarized their research on the synthesis of oxygen-containing compounds via C—H or C—C bond activation with 101 kPa oxygen or air as the green oxidant.[6] In the meanwhile, Saha and Laha et al. summarized the uses of K2S2O8 as a cost-effective inorganic oxidant in metal-catalyzed and metal-free oxidative transformations.[7]
The selectivity control of C—H functionalizations has been regarded as an essential section in the new "Holly Grails" of chemistry.[8] Specific to the category of aromatic C—H bond oxidation, the selectivity mainly originate from electronic effect, steric effect, or chelation assistance. Very recently, several novel reaction systems have been developed for remote C—H bond oxidation. Given all of the above, we feel that it would be helpful to summarize the recent progress in transition metal-catalyzed aromatic C—H oxidations at ortho-, meta-, or para-position.
This review will focus on those aromatic oxidations where the oxygen atom on the newly formed C—O bond should originate from the terminal oxidant (Eq. 1). There are other pathways of aromatic C—H bond functionalizations for C—O bond generations: One way is the reaction between arenes and oxygen-containing nucleophiles via transition metal-catalyzed C—H activations, [9] where the oxidation of transition metal core with an external oxidant is the essential step. It is noteworthy that the anode could also be regarded as an external oxidant, for instance, the groups of Ackermann, Mei, Lei et al. have recently made distinguished contributions to transition metal-catalyzed electrooxidative C—H functionalizations for C—O bond formations.[10] Another way is C—H oxidation via radical aromatic substitution without transition metal catalyst.[11] The above mentioned transformations will not be included in this review.
|
|
(1) |
This review has compiled the research advances of transition metal-catalyzed regio-selective aromatic C—H oxidations up to spring of 2018. We hereby wish to give the readers an intuitive glance at the recent progress of this research area, as well as the generative mechanism of specific chemo- and regio-selectivity.
About 30 years ago, Jintoku, Fujiwara and co-workers reported Pd(OAc)2-catalyzed (OAc=acetate) oxidation of benzene to phenol under the atmosphere of oxygen ((1.52 MPa) and carbon monoxide (1.52 MPa) at 180 ℃. 1, 10-Phenathroline was recognized as the optimized ligand. Under optimized reaction conditions, phenyl acetate was monitored as a major side product, while benzoic acid was isolated as the major product in the absence of ligand.[12]
The above systems have been suffering from high pressure, which could limit the applications of these strategies. In 2007, Ishii and co-workers utilized polyoxometalate H5PMo10V2O40•26H2O (2 mol%) as the redox catalyst in palladium-catalyzed hydroxylation-carboxylation of biphenyl, and the reaction could proceed smoothly under an atmosphere of oxygen (50.7 kPa) and carbon monoxide (50.7 kPa) (Eq. 2).[13]
|
|
(2) |
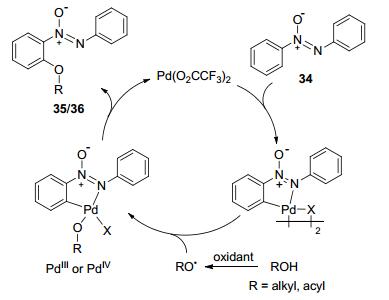
Oxidants other than oxygen have proved applicable to palladium-catalyzed direct aromatic C—H oxidations. In 1996, Crabtree and co-workers employed PhI(OAc)2 as the oxidant in Pd(OAc)2-catalyzed C—H oxidation of arenes, delivering acetoxylated products. Mechanistic studies ruled out a radical route, and indicated that palladation should be the rate-determining step.[14] Later, Sanford and co-workers performed a detailed mechanistic investigation on palladium-catalyzed PhI(OAc)2 oxidized aromatic C—H acetoxylation. The results of NMR and kinetic studies suggested that the most active catalyst rests as a palladium dimer in solution (Scheme 1).[15]
Besides PhI(OAc)2, K2S2O8 has also been used as the terminal oxidant in palladium-catalyzed C—H oxidation. In 2013, Sanford and co-workers reported palladium-catalyzed direct acetoxylation of benzene, using K2S2O8 as the oxidant and a solvent mixture of AcOH/Ac2O.[16] In 2004, Nozaki and co-workers reported the palladium-catalyzed sequential hydroxylation-carboxylation using biphenyl using formic acid as the carbonyl source, generating a mixture of meta- and para-hydroxybiphenylcarboxylic acids in a combined yield of 45% (m-:p-=17:83) (Eq. 3).[17]
|
|
(3) |
In 2013, Guo and co-workers reported tetrakis(pentafluorophenyl)porphyrin manganese(Ⅲ) chloride (F20TPPMnCl) catalyzed hydroxylation of 2-substituted naphthaline (Eq. 4). Depending on the electronic properties of the substituent on C2 position, the catalytic system showed different regio-selectivies. Naphthaline with an electron-donating substituent afforded C1-hydroxylated product, while with an electron-withdrawing substituent predominantly delivered C4-hydroxylated product.[18]
|
|
(4) |
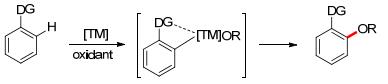
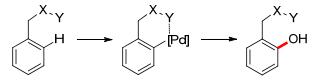
In the category of transition metal-catalyzed ortho-selective aromatic C—H oxidation, the selectivity originated from the coordination of metal catalyst and the Lewis base substituent on the arene, which will tend to form a cyclized transition state via C—H metalation, followed by reductive elimination to afford the ortho-oxygenated product (Scheme 2).
Stoichiometric amount of copper has proved efficacy in the ortho-selective hydroxylation of arenes, using molecular oxygen, [19] iodosylbenzene, [20] hydrogen peroxide, [21] or trimethylamine N-oxide[22] as the terminal oxidant. Besides these copper‑catalyzed aromatic hydroxylations with external oxidants, Cu(OAc)2 itself could also serve as the terminal oxidant in aromatic C—H oxidations. In 1991, Takizawa and co-workers reported stoichiometric amount of Cu(OAc)2 mediated direct ortho-acetoxylation of 4-alkoxylated phenols. The configuration of the product was confirmed by nuclear Overhauser effect (NOE) experiment, and the possibility of transesterification involving the 1-hydroxy group was ruled out. However, this reaction system is not applicable to the acetoxylation of 4-methylphenols and 4-phenylphenols.[23] In 2016, Jana and co-workers reported Cu(OAc)2•H2O mediated hydroxylation via a bidentate directing group consisted of benzamide and 8-aminoquinoline. Radical trap experiment with (2, 2, 6, 6-tetramethylpiperidin-1-yl)oxyl (TEMPO) found no significant effect on the reaction outcome, thus eliminating the radical pathway (Eq. 5).[24]
|
|
(5) |
In 2006, Yu and co-workers observed that an equimolar amount of Cu(OAc)2 could mediate the ortho-hydroxylation of 2-phenylpyridine with 1 equiv of H2O in CH3CN under O2 (101 kPa) at 130 ℃. Labeling experiments using H218O demonstrated that the oxygen atom from Cu(OAc)2 was incorporated into product. The author hypothesized the binding of the hydroxyl group and the nitrogen in the product to Cu(Ⅱ) might prevent catalytic turnover. By adding Ac2O to the reaction system, the copper loading was successfully reduced to 10 mol % under O2 atmosphere, albeit resulting in significant diacetoxylation (Eq. 6).[25]
|
|
(6) |
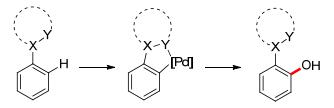
Palladium-catalyzed ortho-C—H oxidation via a 5-membered intermediate is shown in Scheme 3.
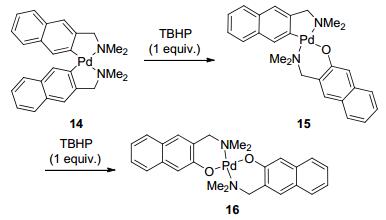
CH2-NR2 as DG In 1990s, Boersma, van Koten, and co-workers studied the generation of heteroleptic diarylpalladium compounds and the oxidation of Pd-Ar bond using tert-butyl hydroperoxide (TBHP) as the terminal oxidant. With the assistance of CH2NR2 group, this transformation regio-selectively delivered mono-hydroxylated compounds (Scheme 4).[26]
Carbonyl group as DG In 2009, Yu and co-workers described the Pd(OAc)2-catalyzed regioselective ortho-hydroxylation of potassium benzoates or benzoic acids, employing the environmentally friendly molecular oxygen as the terminal oxidant (Eq. 7). The presence of benzoquinone (BQ) was found to significantly increase the reaction yield, however, it is not essential. Labeling experiment with 18O2 proved that the oxygen-atom incorporated into the hydroxylated product is originated from molecular oxygen, while oxygen incorporation from H2O was ruled out
|
|
(7) |
via labeling experiment using H218O, indicating a direct oxygenation of the arylpalladium intermediates by molecular oxygen.[27]
In 2012, Rao and co-workers developed a palladiumcatalyzed ortho-hydroxylation system with selectfluor or K2S2O8 as the oxidant and a TFA/TFAA (9:1) mixture as the solvent. This system has proved efficacy toward ketone-, ester-, amide-, acetanilide-, and sulfonamide-directed benzens.[28]
Imine, oxime or azo as DG Imine, oxime or azo as DG is shown in Scheme 5.
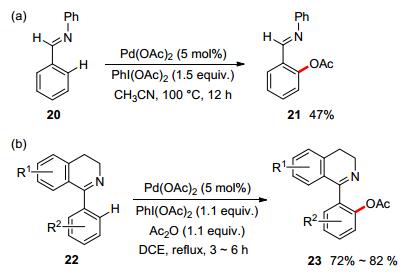
In 2004, Sanford and co-workers proved the feasibility of Pd(OAc)2-catalyzed ortho-selective C—H acetoxylation with imine as the directing group and PhI(OAc)2 as the terminal oxidant, delivering a moderate yield (Scheme 6a).[29] Later, Reddy and co-workers reported the acetoxylation of 1-aryl-3, 4-dihydroisoquinolines under similar reaction conditions devoid of additional stoichiometric amount of Ac2O (Scheme 6b).[30]
Transient directing group (TDG) strategy has recently boomed in transition metal-catalyzed C—H functionalizations.[31, 32] To overcome the weak coordination between formyl group and transition metal, Sorensen and co-workers developed palladium-catalyzed ortho-hydroxylation of arylaldehyde utilizing anthranilic acid as the TDG (Scheme 7). The authors proposed that the reaction might go through a transient imine intermediate with catalytic amount of amines. This catalytic system could work with both electron-donating and electron-withdrawing groups substituted benzaldehydes.[33]
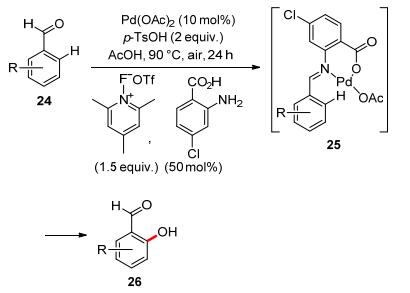
In 2006, Sanford and co-workers reported Pd(OAc)2- catalyzed acetoxylation of arenes with oxime ether as the directing group. They tested various oxidants, and found significant quantities of the ortho-acetoxylated product could be obtained with oxone, K2S2O8, or PhI(OAc)2 as the oxidant (Eq. 8), however, the latter two oxidants could also induce the formation of small amount of di-ortho- acetoxylated product.[34]
|
|
(8) |
With PPh3 as the ligand and CHCl2CHCl2 as the solvent, Jiao and co-workers developed a strategy for Pd(OAc)2- catalyzed ortho-C—H hydroxylation of aryl oxime ethers (Eq. 9).[35] A PdⅡ/PdⅣcatalytic cycle was proposed as follows: firstly, oxime ether assisted ortho-C—H palladation formed a 5-membered palladacycle; secondly, oxidative addition of K2S2O8 with PdⅡ afforded a PdⅣintermediate; finally, reductive elimination gave the hydroxylated product and regenerated PdⅡ catalyst. The ligand was supposed to accelerate the C—O bond reductive elimination step (Scheme 8).
|
|
(9) |
Azobenzene has been known as a functional group in among others Sudan Red B. Moreover, it could be used as the directing group in palladium-catalyzed C—H oxidations since the beginning of this century. In 2004, Sanford and co-workers disclosed Pd(OAc)2-catalyzed azo-assisted ortho-C—H acetoxylation.[29] Later, Joseph and co-workers developed Pd(OAc)2-catalyzed ortho-C—H hydroxylation of symmetrical/unsymmetrical azobenzenes via a PhI- (OTFA)2/DCE system, generating unsymmetrical azophenols (Eq. 10).[36]
|
|
(10) |
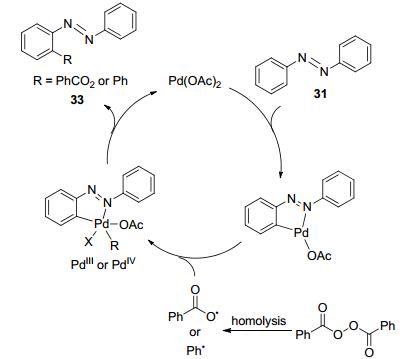
In 2014, Zeng and co-workers reported Pd(OAc)2-catalyzed ortho-esterification of azoarenes using aryl acylperoxide as the oxygen source (Eq. 11). The chemo-selectivity shifted with different solvents and temperatures: C—O functionalized product will be generated with CH3CN as the solvent at 60 ℃, while arylated product will be obtained with PhCl as the solvent at 130 ℃. In contrast with the previously discussed PdⅡ~PdⅣcatalytic cycle, the authors proposed a radical mechanism, which might undergo a PdⅢ or PdⅣ intermediate during the catalytic cycle (Scheme 9).[37]
|
|
(11) |
In 2015, Cui, Wu and co-workers discovered the palladium-catalyzed ortho-esterification/etherification of azoxybenzenes at 100 ℃ or even at ambient temperature (Scheme 10). Based on mechanistic studies, a radical reaction pathway was proposed, including the formation of Pd-substrate complex via ortho-C—H activaition, the generation of oxygen radical and its addition to the palladium center, and reductive elimination to afford the oxygenated product (Scheme 11).[38]
N-Heterocycle as DG Nitrogen-containing heterocycles could be applied as the directing group in palladium-catalyzed ortho-selective aromatic oxidation. In early 2000s, Sanford and co-workers utilized the Pd(OAc)2/ PhI(OAc)2 system for regio-selective C—H oxidation of arenes bearing pyridine or other N-heterocyclic substituents, obtaining mono- or di-oxidized products. Various functional groups are well tolerated, including nitro, fluoride, bromide, and etc.[29, 39] Very recently, Correa and co-workers reported Pd(OAc)2-catalyzed C—H acetoxylation/pivaloxylation of arenes with a triazole directing group, which could be easily prepared via click chemistry.[40] Besides PhI(OAc)2, other oxidants like oxone, TBHP, and H2O2 have also been successfully employed in palladium-catalyzed aromatic C—H oxidation by the group of Kim, [41] Sun, [42] and Itoh, [43] respectively, delivering numerous ortho-hydroxylated products (Eq. 12).
|
|
(12) |
The detailed mechanism of palladium-catalyzed ortho- C—H esterification on 2-phenylpyridine with hypervalent iodine as the oxidant was investigated by Sanford and Ritter, respectively. In 2005, Sanford and co-workers disclosed the PdⅡ-PdⅣcatalytic cycle via mechanistic study. They designed and synthesized a series of stable PdⅣcomplexes and presented detailed study of reductive elimination from this oxidation state which finally afforded oxygenated product (Eq. 13).[44] Later, Sanford and Ritter et al. respectively proposed a PdⅡ-PdⅢ catalytic cycle in palladium-catalyzed C—H oxidation of 2-phenylpyridine as was substantiated by X-ray or kinetic isotope effect studies.[45~47]
|
|
(13) |
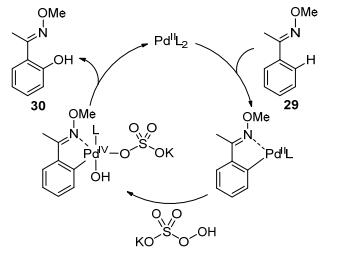
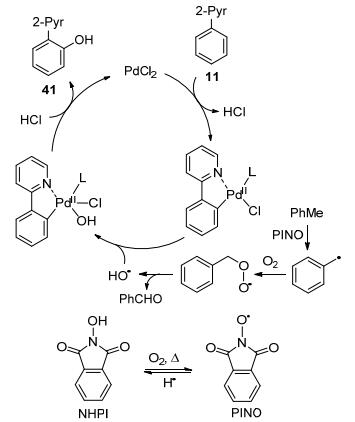
In 2015, Jiao and co-workers reported the palladiumcatalyzed ortho-hydroxylation of 2-phenylpyridine. Interestingly, this system employed catalytic amount of NHPI under oxygen atmosphere (Scheme 12).[48] Isotope labelling strategy proved that the oxygen atom in the hydroxy group originates from molecular oxygen. KIE study (KH/KD=1.86) indicated that C—H bond cleavage might be the rate-limiting step. Based on mechanistic studies, the catalytic cycle was as proposed, the radical stimulation and propagation should follow the sequence NHPI→PINO→ benzyl radical→peroxide radical→hydroxyl radical, which will finally intervened in the palladium catalytic cycle (Scheme 12).
Palladium-catalyzed ortho-C—H oxidation via a 6-membered intermediate is shown in Scheme 13.
CH2-N-heterocycle as DG The diverse CH2-N-heterocycle type directing group-assisted palladium-catalyzed aromatic C—H oxidation was respectively investigated by Sanford and Correa (Eq. 14). With a methylene spacer between the directing group and the aromatic C—H bond, the catalytic cycle would generate a 6-membered palladacycle. Individual kinetic isotope effect experiments were performed with both electron-donating and electron-withdrawing group substituted directing groups. The results demonstrated that reactions could be accelerated by electron-withdrawing groups. A significant kinetic isotope effect was observed, indicating that the formation of the 6-membered palladacycle is the rate determining step.[34, 40, 49]
|
|
(14) |
Anilide or carbamate as DG During the past decades, several groups reported anilide-assisted palladium-catalyzed aromatic C—H oxidation with PhI(O2CR)2, oxone or K2S2O8 as the terminal oxidant, delivering ortho-selective acetoxylated product (Eq. 15).[34, 39, 50, 51] The catalytic system could tolerate various functional groups, including nitro, fluoro, bromo, ketone, and etc.
|
|
(15) |
In 2014, Rao and co-workers reported carbamate-assisted palladium-catalyzed ortho-C—H hydroxylation, using K2S2O8 as the oxidant and a mixture of TFA/TFAA (1:1) as the solvent (Eq. 16). This catalytic system could proceed even at room temperature, however, delivering isolated yields of less than 50%.[52]
|
|
(16) |
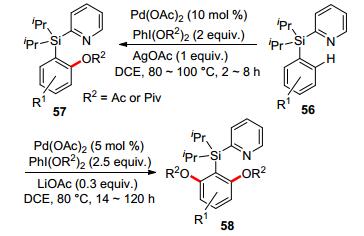
Silicon-bridged DG Silicon-tethered directing groups have been used as easily installable and removable/modifiable auxiliaries for C—H functionalization reactions.[53] In the early 2010s, Gevorgyan and co-workers applied the PyDipSi group as general directing group in palladium-catalyzed ortho-selective mono- or di-oxygenation of arenes, providing access to a variety of acetoxylated and pivaloxylated aromatic compounds in good yields (Scheme 14).[54] It is notable that the palladium-catalyzed oxygenation system not only worked on simple arenes, but also on heterocycles like benzofuran and indole. After the reaction, PyDipSi group could be easily removed or converted into other functional groups.
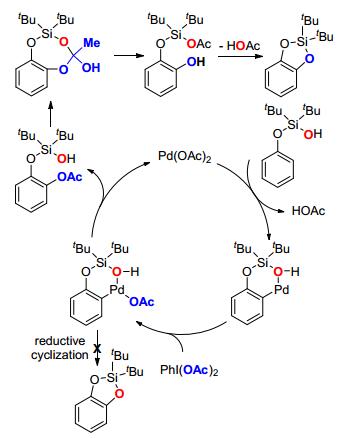
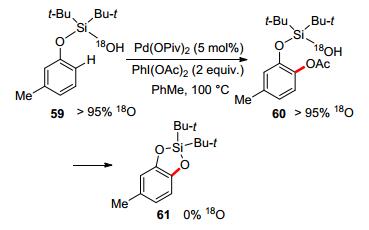
Silanol has also been employed as the directing group in palladium-catalyzed C—H oxidation, delivering a cyclic silicon-protected catechol (Scheme 15). The catechol product could be obtained via a routine desilylation of the silacyle with tetrabutylammonium fluoride (TBAF).[55] To identify the source of oxygen of newly formed aryl silicon ether, labeling experiment was performed with 18O-labelled silanol, after the reaction 18O was detected only in HOAc rather than in the cyclized product. Based on the mechanistic studies, the authors proposed the catalytic cycle, in which reductive cyclization was forbidden, instead, acetoxylation followed by nucleophilic addition delivered the final product (Scheme 16).
Ruthenium-catalyzed ortho-C—H oxidation via a 5-membered intermediate is shown is Scheme 17.
Carbonyl group as DG Recent years have witnessed a tremendous development in the ortho-selective C—H oxidation of arenes and heteroarenes with readily accessible ruthenium catalysts.[5] In 2012, Rao and co-workers investigated [RuCl2(p-cymene)]2-catalyzed ortho-C—H hydroxylation of benzoates. The screening of oxidants demonstrated K2S2O8 and selectfluor to be the optimized oxidants, meanwhile, a TFA/TFAA solvent mixture turned out to be critical. Notably, this catalytic system is valid towards heteroarenes. When using ethyl 1-naphthoate as the substrate, peri-hydroxylated product was found to be predominate isomer rather than ortho-hydroxylated product (Eq. 17).[56]
|
|
(17) |
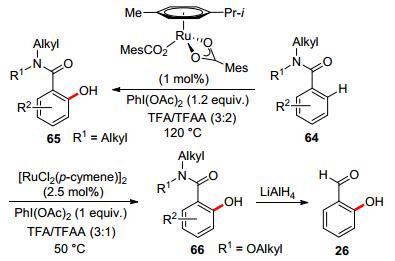
In 2012, Ackermann and co-workers reported ruthenium-catalyzed amide-directed C—H hydroxylation on arenes. Several ruthenium catalysts were examined, out of which the well-defined ruthenium carboxylate complex [Ru(O2CMes)2(p-cymene)][57] was found to be the optimal catalyst at a remarkably low catalyst loading of only 1 mol% (Scheme 18).[58] Later, Weinreb amide was applied as the directing group in ruthenium-catalyzed ortho-C—H hydroxylation of arenes with a similar reaction system but at a milder temperature. In comparison with simple amides, Weinreb amide exhibited more structural flexibility. After reaction, ortho-hydroxylated aryl Weinreb amide could be easily converted into salicylaldehyde.[59]
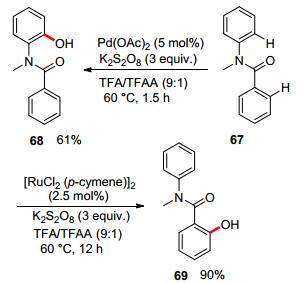
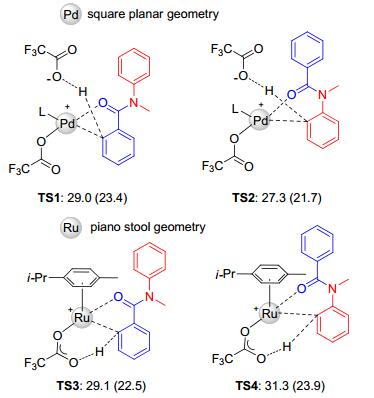
In 2016, Zhang, Rao, and co-workers investigated the C—H oxidation of N-methyl-N-phenylbenzamide using K2S2O8 as the oxidant and TFA/TFAA as the solvent (Scheme 19). It was found that ruthenium and palladium exhibited different regio-selectivities. They rationalized the difference of regio-selectivities to the polarity of transient state via density functional theory (DFT) studies (Figure 1).[51]
In 2012 and 2013, Ackermann and Rao respectively reported ruthenium-catalyzed ortho-C—H bond oxygenations of arenes with weakly coordinating ketones as the directing group.[60, 61] In general, the reaction system employed oxone, K2S2O8, Selectfluor, or PhI(OAc)2 as the terminal oxidant and [RuCl2(p-cymene)]2, [RuCl3(H2O)n] or [Ru(O2CMes)2(p-cymene)] as the catalyst, delivering mono-hydroxylated arylketones or mono/di-hydroxylated diarylketones with ample scope and excellent functional group tolerance. The products could be further converted into various functional molecules. Very recently, flavoneand chromone derivatives have been applied in ruthenium-catalyzed site-selective hydroxylation, again reflecting the potential of this approach in the synthesis of bioactive molecules (Eq. 18).[62]
|
|
(18) |
Aldehyde is supposed to be the weakest carbonyl directing group. It is especially challenging to achieve aldehyde-assisted ortho-hydroxylation due to the inherent tendency of formyl group oxidation under oxidative environment, which will lead to the formation of benzoic acid. In 2014, Ackermann and co-workers developed the [RuCl2(p-cymene)]2/PhI(O2CCF3)2/DCE system which could successfully convert arylaldehyde to salicylaldehyde with high chemo- and regio-selectivities. Intramolecular competition experiments were performed with benzaldehyde bearing an amide, ketone, or ester substituent, indicating that the coordinating ability of aldehyde is weaker than amide, ketone, and ester.63 In contrast, all other transition metals fell short in providing the desired C—H oxygenation (Eq. 19).
|
|
(19) |
|
|
(20) |
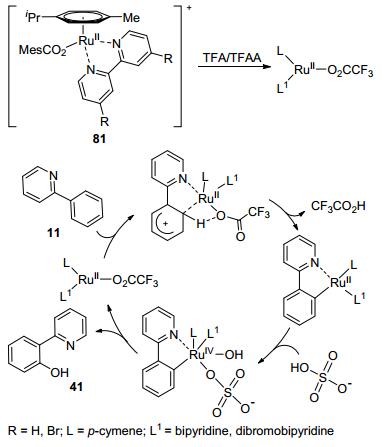
N-Heterocycle as DG Very recently, Singh and co-workers reported ruthenium-catalyzed ortho-hydroxylation of 2-phenylpyridine. The catalysts, naming SPS-Bpy and SPS-DB-Bpy, were derived from [Ru(O2CMes)2(p-cymene)] with bipyridine or dibromobipyridine as the respective substituents. These catalysts were also found to be highly chemo-selective in converting aromatic carboxylate esters. The authors presumed a Ru(Ⅱ)-Ru(Ⅳ) catalytic cycle, including ortho-ruthelation, oxidative formation of C—Ru—O bond, and reductive elimination to afford the final product and regenerate the active catalyst (Scheme 20).[64]
Ruthenium-catalyzed ortho-C—H oxidation via a 6-membered ring transition state is shown in Scheme 21.
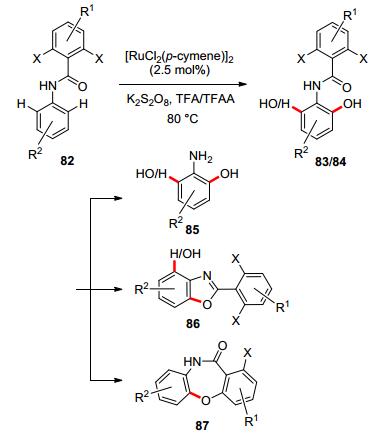
Anilide or carbamate as DG In 2013, Rao and co-workers reported the synthesis of 2-aminophenols and 2-aminobenzene-1, 3-diols via ruthenium-catalyzed ortho-selective mono- or di-hydroxylation with anilide65 as a removable directing group.[66] The reaction demonstrates excellent reactivity, regioselectivity, and good functional group tolerance. Besides removal from hydroxylated products, the ortho-substituted benzoyl group could be further utilized in the synthesis of heterocycles, such as benzoxazoles and dibenzoxazepines (Scheme 22).
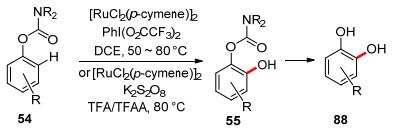
In the same year, Ackermann and co-workers independently reported the application of carbamate as the directing group in ruthenium-catalyzed ortho-hydroxylation. This catalytic system was widely applicable and could efficiently convert para-, meta-, and even more sterically hindered ortho-substituted aryl carbamates bearing various valuable electrophilic functional groups like chloro, bromo, or iodo substituents.[67] Later, Rao and co-workers reported that the ruthenium-catalyzed carbamate assisted C—H hydroxylation system using K2S2O8 as the oxidant and TFA/TFAA as the solvent.[52] It is notable that carbamate directed ortho-hydroxylation is useful for the synthesis of catechol derivatives from easily accessible phenols (Scheme 23).
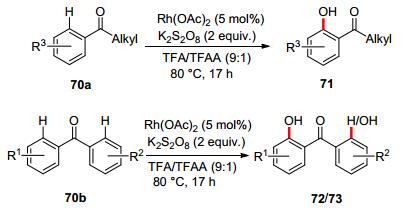
In 2013, Rao and co-workers reported Rh(OAc)2-catalyzed ortho-selective C—H hydroxylation on diarylketones with K2S2O8 as the terminal oxidant and TFA/TFAA (9:1) as the solvent, delivering mono- or 2, 2'-dihydroxylated products in moderate to good yields. This method is also valid for the ortho-hydroxylation of benzoates and benzamides (Scheme 24).[61]
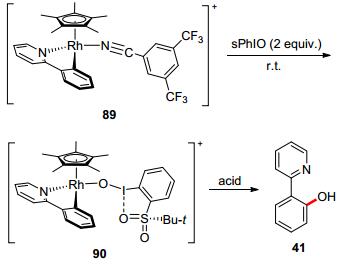
To understand the detailed mechanism of rhodium-catalyzed aromatic C—H oxidation, Jones, Templeton, and co-workers studied the oxygen insertion into the Cp*-Rh-bpp half-sandwich complex using 2-tert-butylsulfonyliodosylbenzene (sPhIO) as the oxidant, forming a Rh(Ⅳ) complex. Treatment with acid generated 2-pyridylphenol (Scheme 25).[68]
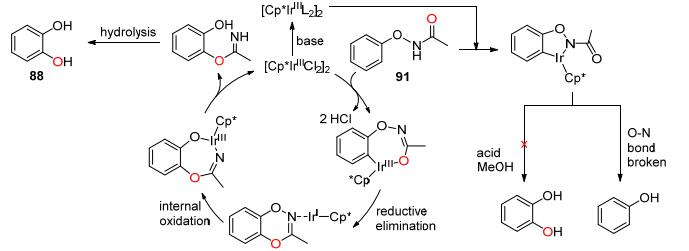
In 2017, Zhao and co-workers developed a redox-neutral strategy for catechol synthesis via iridium-catalyzed ortho-hydroxylation with an internal oxidant.[69] The reaction was performed at room temperature and could tolerate various functional groups like halogens, carboxylates, heterocycles, and even chiral substituents. To understand the oxygen source, isotope labelling experiments were carried out, demonstrating an internal oxidant pathway. The proposed catalytic cycle started from iridium-catalyzed ortho-C—H activation, forming a 7-membered transition state, which underwent reductive elimination to form the C—O bond, oxidative insertion of IrI (Scheme 26) into the N—O bond and subsequently protonation afforded the original IrⅢ catalyst and the imine compound, which would be converted to catechols via hydrolysis.
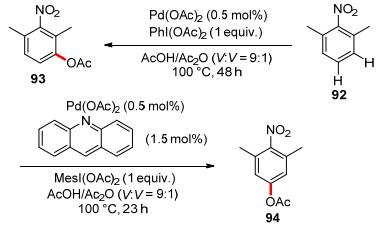
In 2013, Sanford and co-workers disclosed that the regio-selectivity of palladium-catalyzed C—H acetoxylation could be altered with acridine as the ancillary ligand and MesI(OAc)2 as the terminal oxidant, delivering meta-acetoxylated arene (Scheme 27), thus overrided the substrate electronic bias that dominates the regio-selectivity observed in the reaction that used ligand-free Pd(OAc)2 as the catalyst and PhI(OAc)2 as the oxidant.[70]
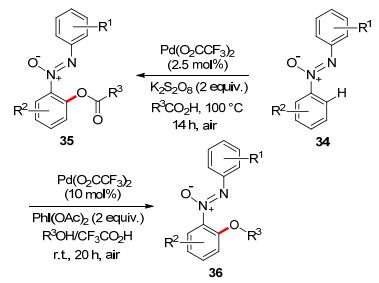
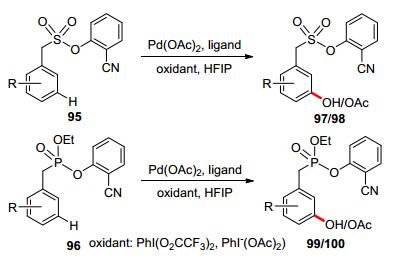
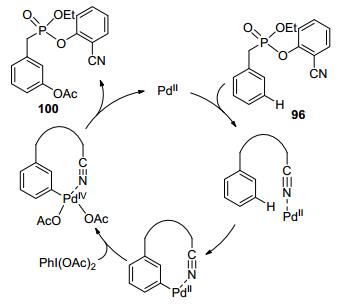
Very recently, template-directed meta-selective C(sp2)— H functionalization has been recognized as a powerful toolin synthetic chemistry.[71] In the category of transition metal-catalyzed meta-selective C—H oxidations, palladium-catalyzed meta-selective C—H oxygenation has been recently achieved with the assistance of a U-shaped template bearing a sulfonate or phosphonate linkage. The catalytic system has exhibited excellent meta-selectivity, delivering mono-hydroxylated/acetoxylated products in high yields (Scheme 28).[72]
Based on mechanistic studies, the author proposed a PdⅡ/PdⅣcatalytic cycle: Firstly, the palladium catalyst coordinated with cyanide group, followed by the meta-C—H activation to form an 11-membered ring transition state. PdⅡ was then oxidized to PdⅣby hypervalent iodine reagent. Finally, reductive elimination delivered the oxidized product and regenerated PdⅡ catalyst (Scheme 29).
The ruthenium-catalyzed para-C—H oxidation without a Lewis base directing group was achieved by Ackermann and co-workers.[67] Taking advantage of the [RuCl2(p-cy-mene)]2/PhI(O2CCF3)2/DCE system, anisole derivatives were converted to the corresponding para-hydroxylated phenols. Oxidations of anisole in the presence of catalytic (10 mol%) or stoichiometric (1 equiv.) amounts of TEMPO furnished product in significantly reduced yields of 43% and 5%, respectively, indicating a single-electrontransfer (SET) pathway. However, the ruthenium catalyst's exact mode of action is as of yet not completely understood (Eq. 21).
|
|
(21) |
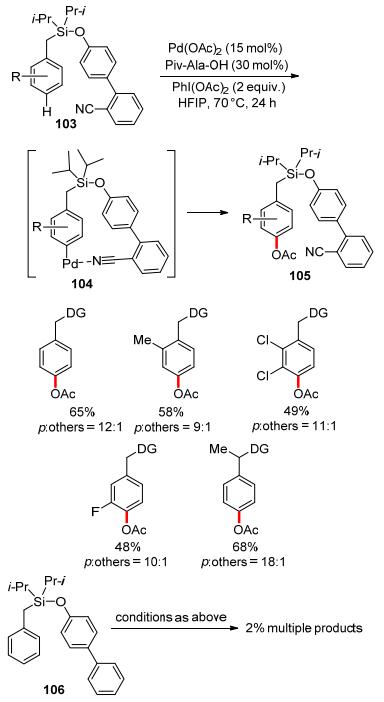
In 2015, Maiti and co-workers disclosed the palladium-catalyzed para-acetoxylation of substituted toluene derivatives (Scheme 30).[73, 74] High para-selectivity was achieved with the assistance of a U-shaped template. Installing two sterically hindered isopropyl groups at the Si atom might allow closer approach of the coordinating group to the para-C—H bond via the Thorpe-Ingold effect. In the control experiment without cyanide group, no para-acetoxylated product was generated.
Recent years have witnessed tremendous progress in transition metal-catalyzed aromatic C—H oxidations. Enormous amount of phenols and phenolic derivatives have been synthesized via this strategy, in the meantime, huge advances have been made in the mechanistic study of these transformations. However, there are still many challenges to be addressed, such as developing cost-competitive transition metal catalysts, exploring switchable directing groups, expanding substrate scope, and reducing reaction temperature. In addition, there are two points deserve particular attention:
Point 1. Chelation-assisted aromatic C—H oxidation has been comprehensively studied over the past few years, delivering ortho-functionalized product. However, there have been only a few examples of meta- and para-selective aromatic C—H oxidations. Considering the diversity of functional molecules, the developing of synthetic methodologies for meta- and para-hydroxylated arenes are undoubtedly in great demand.
Point 2. Most of the oxidants are generally expensive and may cause environmental risks. The utilization of inexpensive peroxides, oxygen or even air as a green oxidant in transition metal-catalyzed aromatic C—H oxidations is more advantageous, thus worth further research. However, the usually used high pressure reaction system could limit the development of this strategy. An additional redox catalyst might be the key solution to facilitate the reaction at ambient atmospheric pressure.
(a) Rappoport, Z. The Chemistry of Phenols, Wiley-VCH, Weinheim, 2003.
(b) Fiegel, H.; Voges, H. W.; Hamamoto, T.; Umemura, S.; Iwata, T.; Miki, H.; Fujita, Y.; Buysch, H. J.; Garbe, D.; Paulus, W. Phenol Derivatives in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, New York, 2002.
(c) Hartwig, J. F. In Handbook of Organopalladium Chemistry for Organic Synthesis, Vol. 1, Ed.: Negishi, E.-I., Wiley-Interscience, New York, 2002, p. 1097.
(d) Tyman, J. H. P. Synthetic and Natural Phenols, Elsevier, New York, 1996.
(e) Liao, S.; Zhang, G.; Cao, H.; Chen, B.; Li, W.; Wu, X.; Feng, Y. Chin. J. Org. Chem. 2018, 38, 1549.
(f) Lin, W.; Cai, Q.; Zheng, C.; Huang, Z.; Shi, D. Chin. J. Org. Chem. 2017, 37, 2094.
(g) Li, Z.; Kang, S.; Chen, L.; Wang, Y.; Li, J. Chin. J. Org. Chem. 2016, 36, 1143.
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147. doi: 10.1021/cr900184e
Enthaler, S.; Company, A. Chem. Soc. Rev. 2011, 40, 4912. doi: 10.1039/c1cs15085e
Thirunavukkarasu, V. S.; Kozhushkov, S. I.; Ackermann, L. Chem. Commun. 2014, 50, 29. doi: 10.1039/C3CC47028H
Newhouse, T.; Baran, P. S. Angew. Chem. Int. Ed. 2011, 50, 3362. doi: 10.1002/anie.201006368
Liang, Y.-F.; Jiao, N. Acc. Chem. Res. 2017, 50, 1640. doi: 10.1021/acs.accounts.7b00108
Mandal, S.; Bera, T.; Dubey, G.; Saha, J.; Laha, J. K. ACS Catal. 2018, 8, 5085. doi: 10.1021/acscatal.8b00743
(a) Huang, Z.; Dong, G. Acc. Chem. Res. 2017, 50, 465.
(b) Hartwig, J. F. Acc. Chem. Res. 2017, 50, 549.
(c) Kozlowski, M. C. Acc. Chem. Res. 2017, 50, 638.
For selected recent examples, see:
(a) Raghuvanshi, K.; Zell, D.; Ackermann, L. Org. Lett. 2017, 19, 1278.
(b) Raghuvanshi, K.; Rauch, K.; Ackermann, L. Chem.-Eur. J. 2015, 21, 1790.
(c) Hao, X.-Q.; Chen, L.-J.; Ren, B.; Li, L.-Y.; Yang, X.-Y.; Gong, J.-F.; Niu, J.-L.; Song M.-P. Org. Lett. 2014, 16, 1104.
(d) Shi, S.; Kuang, C. J. Org. Chem. 2014, 79, 6105.
(e) Zhang, C.; Sun, P. J. Org. Chem. 2014, 79, 8457.
(f) Péron, F.; Fossey, C.; Sopkova-de Oliveira Santos, J.; Cailly, T.; Fabis, F. Chem.-Eur. J. 2014, 20, 7507.
(g) Bhadra, S.; Dzik, W. I.; Gooßen, L. J. Angew. Chem., Int. Ed. 2013, 52, 2959.
(h) Bhadra, S.; Matheis, C.; Katayev, D.; Gooßen, L. J. Angew. Chem., Int. Ed. 2013, 52, 9279.
(i) Suess, A. M.; Ertem, M. Z.; Cramer, C. J.; Stahl, S. S. J. Am. Chem. Soc. 2013, 135, 9797.
(j) Roane, J.; Daugulis, O. Org. Lett. 2013, 15, 5842.
(k) Eom, D.; Jeong, Y.; Kim, Y. R.; Lee, E.; Choi, W.; Lee, P. H. Org. Lett. 2013, 15, 5210.
(l) Zhao, J.; Wang, Y.; He, Y.; Liu, L.; Zhu, Q. Org. Lett. 2012, 14, 1078.
(m) Zhao, J.; Zhang, Q.; Liu, L.; He, Y.; Li, J.; Li, J.; Zhu, Q. Org. Lett. 2012, 14, 5362.
(n) Li, W.; Sun, P. J. Org. Chem. 2012, 77, 8362.
(o) Jiang, T.-S.; Wang, G.-W. J. Org. Chem. 2012, 77, 9504.
(p) Xiao, B.; Gong, T.-J.; Liu, Z.-J.; Liu, J.-H.; Luo, D.-F.; Xu, J.; Liu L. J. Am. Chem. Soc. 2011, 133, 9250.
(q) Anand, M.; Sunoj, R. B. Org. Lett. 2011, 13, 4802.
(r) Wei, Y.; Yoshikai, N. Org. Lett. 2011, 13, 5504.
(s) Wang, X.; Lu, Y.; Dai, H.-X.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 12203.
(t) Wang, G.-W.; Yuan, T.-T. J. Org. Chem. 2010, 75, 476.
For selected reviews and recent examples, see:
(a) Tang, S.; Liu, Y.; Lei, A. Chem 2018, 4, 27.
(b) Tang, S.; Zeng, L.; Lei, A. J. Am. Chem. Soc. 2018, 140, 13128.
(c) Ma, C.; Fang, P.; Mei, T.-S. ACS Catal. 2018, 8, 7179.
(d) Sauermann, N.; Meyer, T. H.; Qiu, Y.; Ackermann, L. ACS Catal. 2018, 8, 7086.
(e) Sauermann, N.; Meyer, T. H.; Ackermann, L. Chem.-Eur. J. 2018, 24, 16209.
(f) Jiao, K.-J.; Li, Z.-M.; Xu, X.-T.; Zhang, L.-P.; Li, Y.-Q.; Zhang, K.; Mei, T.-S. Org. Chem. Front. 2018, 5, 2244.
(g) Shao, A.; Li, N.; Gao, Y.; Zhan, J.; Chiang, C.-W.; Lei, A. Chin. J. Chem. 2018, 36, 619.
(h) Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338.
(i) Jiao, K.-J.; Zhao, C.-Q.; Fang, P.; Mei, T.-S. Tetrahedron Lett. 2017, 58, 797.
(j) Yuan, Y.; Chen, Y.; Tang, S.; Huang, Z.; Lei, A. Sci. Adv. 2018, 4, eaat5312.
(k) Shrestha, A.; Lee, M.; Dunn, A. L.; Sanford, M. S. Org. Lett. 2018, 20, 204.
(l) Sauermann, N.; Meyer, T. H.; Tian, C.; Ackermann, L. J. Am. Chem. Soc. 2017, 139, 18452.
(m) Yang, Q.-L.; Li, Y.-Q.; Ma, C.; Fang, P.; Zhang, X.-J.; Mei, T.-S. J. Am. Chem. Soc. 2017, 139, 3293.
(n) Li, Y.-Q.; Yang, Q.-L.; Fang, P.; Mei, T.-S.; Zhang, D. Org. Lett. 2017, 19, 2905.
A recent example: Zhang, Z.-J.; Quan, X.-J.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Org. Lett. 2014, 16, 3292.
(a) Jintoku, T.; Taniguchi, H.; Fujiwara, Y. Chem. Lett. 1987, 1865.
(b) Jintoku, T.; Takaki, K.; Fujiwara, Y.; Fuchita Y.; Hiraki, K. Bull. Chem. Soc. Jpn. 1990, 63, 438.
(c) Jintoku, T.; Nishimura, K.; Takaki, K.; Fujiwara, Y. Chem. Lett. 1990, 1687.
Yamada, S.; Sakaguchi, S.; Ishii, Y. J. Mol. Catal. A Chem. 2007, 262, 48. doi: 10.1016/j.molcata.2006.08.045
Yoneyama, T.; Crabtree, R. H. J. Mol. Catal. A: Chem. 1996, 108, 35. doi: 10.1016/1381-1169(95)00289-8
Cook, A. K.; Sanford, M. S. J. Am. Chem. Soc. 2015, 137, 3109. doi: 10.1021/jacs.5b00238
Gary, J. B.; Cook, A. K.; Sanford, M. S. ACS Catal. 2013, 3, 700. doi: 10.1021/cs300786j
Shibahara, F.; Kinoshita, S.; Nozaki, K. Org. Lett. 2004, 6, 2437. doi: 10.1021/ol049166l
Chen, C.-D.; Sheng, W.-B.; Shi, G.-J.; Guo, C.-C. J. Phys. Org. Chem. 2013, 26, 23. doi: 10.1002/poc.v26.1
Casella, L.; Rigoni, L. J. Chem. Soc., Chem. Commun. 1985, 1668. https://pubs.rsc.org/en/content/articlelanding/1985/c3/c39850001668/unauth#!divAbstract
Réglier, M.; Amadeï, E.; Tadayoni, R.; Waegell, B. J. Chem. Soc., Chem. Commun. 1989, 447. https://pubs.rsc.org/en/content/articlelanding/1989/c3/c39890000447/unauth#!divAbstract
Cruse, R. W.; Kaderli, S.; Meyer, C. J.; Zuberbühler, A. D.; Karlin, K. D. J. Am. Chem. Soc. 1988, 110, 5020. doi: 10.1021/ja00223a020
Reinaud, O.; Capdevielle, P.; Maumy, M. J. Chem. Soc., Chem. Commun. 1990, 566. https://pubs.rsc.org/en/content/articlelanding/c3/1990/c39900000566#!divAbstract
Takizawa, Y.; Tateishi, A.; Sugiyama, J.; Yoshida, H.; Yoshihara, N. J. Chem. Soc., Chem. Commun. 1991, 104. doi: 10.1002/chin.199122108
Singh, B. K.; Jana, R. J. Org. Chem. 2016, 81, 831. doi: 10.1021/acs.joc.5b02302
Chen, X.; Hao, X.-S.; Goodhue, C.-E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 6790. doi: 10.1021/ja061715q
(a) Valk, J.-M.; van Belzen, R.; Boersma, J.; Spek, A. L.; van Koten, G. J. Chem. Soc., Dalton Trans. 1994, 2293.
(b) Valk, J.-M.; Boersma, J.; van Koten, G. Organometallics 1996, 15, 4366.
Zhang, Y.-H.; Yu, J.-Q. J. Am. Chem. Soc. 2009, 131, 14654. doi: 10.1021/ja907198n
Shan, G.; Yang, X.; Ma, L.; Rao, Y. Angew. Chem., Int. Ed. 2012, 51, 13070. doi: 10.1002/anie.201207458
Dick, A. R.; Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2004, 126, 2300. doi: 10.1021/ja031543m
Subba Reddy, B. V.; Umadevi, N.; Narasimhulu, G.; Yadav, J. S. Tetrahedron Lett. 2012, 53, 6091. doi: 10.1016/j.tetlet.2012.08.124
Zhao, Q.; Poisson, T.; Pannecoucke, X.; Besset, T. Synthesis 2017, 49, 4808. doi: 10.1055/s-0036-1590878
Gandeepan, P.; Ackermann, L. Chem 2018, 4, 199. doi: 10.1016/j.chempr.2017.11.002
Chen, X.-Y.; Ozturk, S.; Sorensen, E. J. Org. Lett. 2017, 19, 6280. doi: 10.1021/acs.orglett.7b02906
Desai, L. V.; Malik, H. A.; Sanford, M. S. Org. Lett. 2006, 8, 1141. doi: 10.1021/ol0530272
Liang, Y.-F.; Wang, X.; Yuan, Y.; Liang, Y.; Li, X.; Jiao, N. ACS Catal. 2015, 5, 6148. doi: 10.1021/acscatal.5b01700
Nguyen, T. H. L.; Gigant, N.; Delarue-Cochin, S.; Joseph, D. J. Org. Chem. 2016, 81, 1850. doi: 10.1021/acs.joc.5b02614
Qian, C.; Lin, D.; Deng, Y.; Zhang, X.-Q.; Jiang, H.; Miao, G.; Tang, X.; Zeng, W. Org. Biomol. Chem. 2014, 12, 5866. doi: 10.1039/C4OB00993B
Zhang, D.; Cui, X.; Yang, F.; Zhang, Q.; Zhu, Y.; Wu, Y. Org. Chem. Front. 2015, 2, 951. doi: 10.1039/C5QO00120J
Kalyani, D.; Sanford, M. S. Org. Lett. 2005, 7, 4149. doi: 10.1021/ol051486x
Irastorza, A.; Aizpurua, J. M.; Correa, A. Org. Lett. 2016, 18, 1080. doi: 10.1021/acs.orglett.6b00195
Kim, S. H.; Lee, H. S.; Kim, S. H.; Kim, J. N. Tetrahedron Lett. 2008, 49, 5863. doi: 10.1016/j.tetlet.2008.07.141
Dong, J.; Liu, P.; Sun, P. J. Org. Chem. 2015, 80, 2925. doi: 10.1021/acs.joc.5b00167
Yamaguchi, T.; Yamaguchi, E.; Tada, N.; Itoh, A. Adv. Synth. Catal. 2015, 357, 2017. doi: 10.1002/adsc.v357.9
Dick, A. R.; Kampf, J. W.; Sanford, M. S. J. Am. Chem. Soc. 2005, 127, 12790. doi: 10.1021/ja0541940
Stowers, K. J.; Sanford, M. S. Org. Lett. 2009, 11, 4584. doi: 10.1021/ol901820w
Powers, D. C.; Geibel, M. A. L.; Klein, J. E. M. N.; Ritter, T. J. Am. Chem. Soc. 2009, 131, 17050. doi: 10.1021/ja906935c
Powers, D. C.; Xiao, D. Y.; Geibel, M. A. L.; Ritter, T. J. Am. Chem. Soc. 2010, 132, 14530. doi: 10.1021/ja1054274
Yan, Y.; Feng, P.; Zheng, Q.-Z.; Liang, Y.-F.; Lu, J.-F.; Cui, Y.; Jiao, N. Angew. Chem., Int. Ed. 2013, 52, 5827. doi: 10.1002/anie.201300957
Desai, L. V; Stowers, K. J.; Sanford, M. S. J. Am. Chem. Soc. 2008, 130, 13285. doi: 10.1021/ja8045519
Wang, G.-W.; Yuan, T.-T.; Wu, X.-L. J. Org. Chem. 2008, 73, 4717. doi: 10.1021/jo8003088
Sun, Y.-H.; Sun, T.-Y.; Wu, Y.-D.; Zhang, X.; Rao, Y. Chem. Sci. 2016, 7, 2229. doi: 10.1039/C5SC03905C
Yang, X.; Sun, Y.; Chen, Z.; Rao, Y. Adv. Synth. Catal. 2014, 356, 1625. doi: 10.1002/adsc.v356.7
A review: Parasram, M.; Gevorgyan, V. Acc. Chem. Res. 2017, 50, 2038.
(a) Chernyak, N.; Dudnik, A. S.; Huang, C.; Gevorgyan, V. J. Am. Chem. Soc. 2010, 132, 8270.
(b) Huang, C.; Chernyak, N.; Dudnik, A. S.; Gevorgyan, V. Adv. Synth. Catal. 2011, 353, 1285.
(c) Gulevich, A. V.; Melkonyan, F. S.; Sarkar, D.; Gevorgyan, V. J. Am. Chem. Soc. 2012, 134, 5528.
(a) Huang, C.; Ghavtadze, N.; Chattopadhyay, B.; Gevorgyan, V. J. Am. Chem. Soc. 2011, 133, 17630.
(b) Huang, C.; Ghavtadze, N.; Godoi, B.; Gevorgyan, V. Chem.-Eur. J. 2012, 18, 9789.
Yang, Y.; Lin, Y.; Rao, Y. Org. Lett. 2012, 14, 2874. doi: 10.1021/ol301104n
Ackermann, L.; Vicente, R.; Potukuchi, H. K.; Pirovano, V. Org. Lett. 2010, 12, 5032. doi: 10.1021/ol102187e
Thirunavukkarasu, V. S.; Hubrich, J.; Ackermann, L. Org. Lett. 2012, 14, 4210. doi: 10.1021/ol3018819
Yang, F.; Ackermann, L. Org. Lett. 2013, 15, 718. doi: 10.1021/ol303520h
Thirunavukkarasu, V. S.; Ackermann, L. Org. Lett. 2012, 14, 6206. doi: 10.1021/ol302956s
Shan, G.; Han, X.; Lin, Y.; Yu, S.; Rao, Y. Org. Biomol. Chem. 2013, 11, 2318. doi: 10.1039/c3ob27457h
Kim, K.; Choe, H.; Jeong, Y.; Lee, J. H.; Hong, S. Org. Lett. 2015, 17, 2550. doi: 10.1021/acs.orglett.5b01138
Yang, F.; Rauch, K.; Kettelhoit, K.; Ackermann, L. Angew. Chem., Int. Ed. 2014, 53, 11285. doi: 10.1002/anie.201405647
Shome, S.; Singh, S. P. Tetrahedron Lett. 2017, 58, 3743. doi: 10.1016/j.tetlet.2017.08.037
Ackermann, L.; Wang, L.; Wolfram, R.; Lygin, A. V. Org. Lett. 2012, 14, 728. doi: 10.1021/ol203251s
Yang, X.; Shan, G.; Rao, Y. Org. Lett. 2013, 15, 2334. doi: 10.1021/ol400437a
Liu, W.; Ackermann, L. Org. Lett. 2013, 15, 3484. doi: 10.1021/ol401535k
Turlington, C. R.; Morris, J.; White, P. S.; Brennessel, W. W.; Jones, W. D.; Brookhart, M.; Templeton, J. L. Organometallics 2014, 33, 4442. doi: 10.1021/om500660n
Wu, Q.; Yan, D.; Chen, Y.; Wang, T.; Xiong, F.; Wei, W.; Lu, Y.; Sun, W.-Y.; Li, J. J.; Zhao, J. Nat. Commun. 2017, 8, 14227. doi: 10.1038/ncomms14227
Cook, A. K.; Emmert, M. H.; Sanford, M. S. Org. Lett. 2013, 15, 5428. doi: 10.1021/ol4024248
For selected recent views on template-directed meta-selective C(sp2)—H functionalizations, see:
(a) Mihai, M. T.; Genov, G. R.; Phipps, R. J. Chem. Soc. Rev. 2017, 47, 149.
(b) Ping, L.; Chung, D. S.; Bouffard, J.; Lee, S.-G. Chem. Soc. Rev. 2017, 46, 4299.
(c) Mihai, M. T.; Phipps, R. J. Synlett 2017, 28, 1011.
(d) Dey, A.; Agasti, S.; Maiti, D. Org. Biomol. Chem. 2016, 14, 5440.
(e) Yang, G.; Butt, N.; Zhang, W. Chin. J. Catal. 2016, 37, 98.
(f) Yang, G.; Butt, N.; Zhang, W. Chin. J. Catal. 2016, 37, 98.
(g) Ackermann, L.; Li, J. Nat. Chem. 2015, 7, 686.
(h) Yang, J. Org. Biomol. Chem. 2015, 13, 1930.
(a) Bera, M.; Sahoo, S. K.; Maiti, D. ACS Catal. 2016, 6, 3575.
(b) Maji, A.; Bhaskararao, B.; Singha, S.; Sunoj, R. B.; Maiti, D. Chem. Sci. 2016, 7, 3147.
Bag, S.; Patra, T.; Modak, A.; Deb, A.; Maity, S.; Dutta, U.; Dey, A.; Kancherla, R.; Maji, A.; Hazra, A.; Bera, M.; Maiti, D. J. Am. Chem. Soc. 2015, 137, 11888. doi: 10.1021/jacs.5b06793
A review on template-directed para-selective C(sp2)–H functionalizations: Dey, A.; Maity, S.; Maiti, D. Chem. Commun. 2016, 52, 12398.

Scheme 1 Proposed catalytic cycle of palladium-catalyzed PhI- (OAc)2 oxidized aromatic C—H acetoxylation

Scheme 7 Pd(OAc)2-catalyzed ortho-C—H hydroxylation of arylaldehyde via a TDG strategy

Scheme 8 Proposed catalytic cycle of Pd(OAc)2-catalyzed ortho-hydroxylation of aryl oxime ethers

Scheme 9 Proposed catalytic cycle of Pd(OAc)2-catalyzed ortho-esterification of azoarenes with aryl acylperoxide

Scheme 11 Proposed catalytic cycle of Pd(O2CCF3)2-catalyzed ortho-oxidation of azoxybenzenes

Scheme 12 Palladium-catalyzed ortho-hydroxylation of 2-phenylpyridine with NHPI as the co-catalyst

Scheme 16 Proposed mechanism of palladium-catalyzed synthesis of cyclic silicon-protected catechol

Scheme 19 Palladium-/ruthenium-catalyzed ortho-C—H hydroxylation of N-methyl-N-phenylbenzamide

Scheme 20 Proposed catalytic cycle of ortho-hydroxylation of 2-phenylpyridine with 81 as the pre-catalyst

Scheme 21 Ruthenium-catalyzed ortho-C—H oxidation via a 6-membered ring transition state

Scheme 26 Proposed catalytic cycle of iridium-catalyzed catechol synthesis with internal oxidant

Scheme 27 Ligand-controlled regio-selectivity shift of palladium-catalyzed aromatic acetoxylation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



















 下载:
下载:


















