Received Date:
24 March 2020 Available Online:
15 June 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (No. 21975003) and the Program for Innovative Research Team in Anqing Normal University
Abstract:
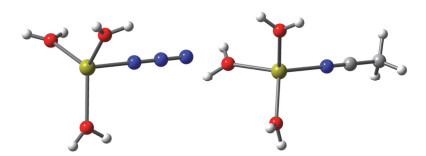
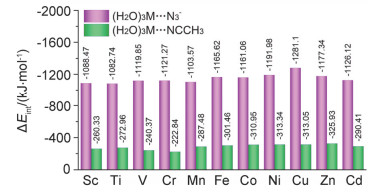
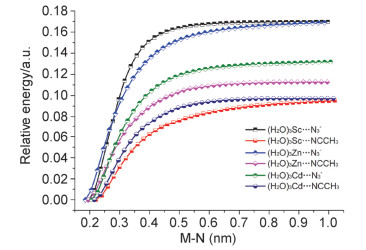
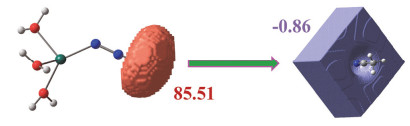
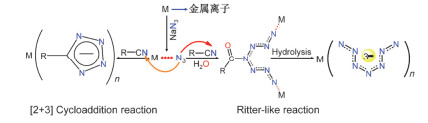
The study on the reaction mechanism of organonitrile and sodium azide catalyzed by transition metals has always been a challenging and controversial task. Due to the difficulty in capturing the reaction intermediates, there is still no direct evidence to uncover the nature of the reaction. In this paper, the reaction mechanism has been explored by using a combining theoretical and experimental method. Based on the theoretical analysis of the stability of two types of intermediates (H2O)3M…N3- and (H2O)3M…NCCH3 and the successful capture of two activated intermediates containing metal cadmium ions Cd2(μ3-N3)(μ3-OH)(μ5-CHDA) (1) and Cd(μ2-N3)(μ3-IBA) (2) (H2CHDA=1, 3-cycloadipic acid and HIBA=4-(imi-dazol-1-yl) benzoic acid), which were achieved under the hydrothermal conditions and characterized by single-crystal XRD analysis. For the first time, the experimental and theoretical results reveal that the transition metal ions activate the azide rather than the cyano group of nitriles. In addition, the results of both the electrostatic potential basins analysis of activated intermediates (H2O)3M…N3- and acetonitrile molecules obtained by the theoretical calculation and our recently reported experimental results reveal that the intermediates (H2O)3M…N3- can be used as electrophilic reagent. Its uncoordinated terminal N atom can attack the N atom of the cyano group of acetonitrile to undergo a nucleophilic addition reaction during the chemical reaction progress, and then it may undergo a[2+3] cycloaddition reaction to in-situ form tetrazole. Moreover, with the aid of water molecules, its adducts may also occur similar to the Ritter-like reaction to in-situ form polynitrogen anion. Our findings may open a novel field of the in-situ synthesis of polynitrogen compounds based on the transition metal-catalyzed reactions of organonitrile and azide.
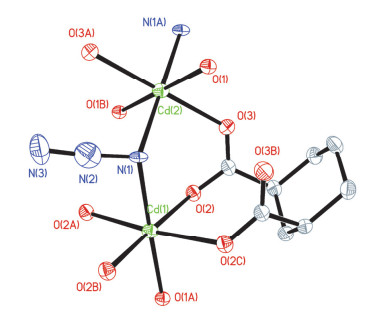
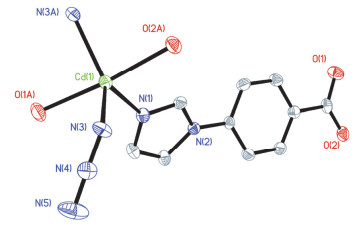
Figure 7.
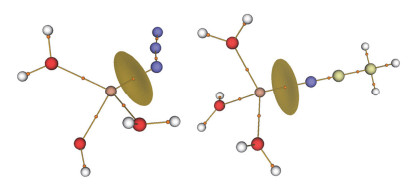
Coordination environments of the Cd2+centers in 1 with the ellipsoids drawn at the 30% probability level, all hydrogen atoms are omitted for clarity.
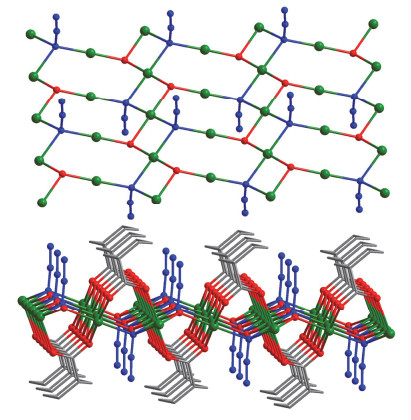
Figure 10.
Coordination environments of the Cd2+centers in 2 with the ellipsoids drawn at the 30% probability level, all hydrogen atoms are omitted for clarity
Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. J. Chem. Phys. 2004, 387, 481.
[24]
Ritter, J. J.; Minieri, P. P. J. Am. Chem. Soc. 1948, 70, 4045. doi: 10.1021/ja01192a022
[25]
Ritter, J. J.; Kalish, J. J. Am. Chem. Soc. 1948, 70, 4048. doi: 10.1021/ja01192a023
[26]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013.
Figure 7
Coordination environments of the Cd2+centers in 1 with the ellipsoids drawn at the 30% probability level, all hydrogen atoms are omitted for clarity.
Figure 10
Coordination environments of the Cd2+centers in 2 with the ellipsoids drawn at the 30% probability level, all hydrogen atoms are omitted for clarity

 下载:
下载:


 下载:
下载:














