表 1
氨基噁唑啉呫吨类BACE1抑制剂的结构和生物活性
Table 1.
Structure and biological activity of aminooxazoline xanthene BACE1 inhibitors

阿尔茨海默症(AD)是一种具有致命威胁的慢性神经退行性疾病,是导致痴呆的主要原因,目前尚无有效治疗方法[1]。AD的主要病理学特征是大脑中β-淀粉样蛋白(Aβ)的过量沉积,由β-分泌酶(BACE1)和γ-分泌酶依次切割β-淀粉样蛋白前体蛋白(APP)而产生[2]。因此,BACE1作为APP代谢过程中的初始关键酶,已成为治疗AD疾病的潜在靶点,BACE1抑制剂的开发是目前AD治疗的重要方向[2]。早期开发的BACE1抑制剂主要以拟肽类化合物为主,渗透性和口服生物利用度差,类药特性不明显[3]。因此,近几年发展的BACE1抑制剂多为小分子化合物。在目前涌现的数以万计的BACE1抑制剂中[3],进入临床试验阶段的却寥寥无几,而且能够通过临床试验的抑制剂至今尚未见报道[4]。可见,新药的研发至今还是一件耗费巨大且效率低下的工作。计算机辅助药物设计技术为新型BACE1抑制剂的设计和合成提供了强有力的理论支持,已广泛应用于临床药物的开发。该方法根据与疾病相关的靶标分子的三维结构来定向设计结构、性质互补的小分子配体,大大降低新药研发成本,缩短周期[5]。
最近发现,由多个研究小组开发的氨基噁唑啉呫吨类化合物表现出了较强的BACE1抑制活性,类药特性明显[6, 7]。本文采用计算机辅助药物设计中广泛应用的比较分子相似性指数(CoMSIA)和分子对接方法模拟分析氨基噁唑啉呫吨类化合物的构效关系,寻找影响分子活性的特征结构,揭示小分子配体与蛋白质受体间的作用机制[8],为新型高效抑制剂的设计和改造提供理论指导,提高研发成功率。
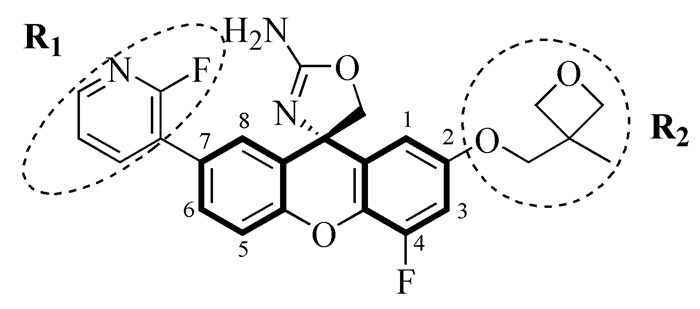
66个氨基噁唑啉呫吨类BACE1抑制剂来自于Epstein和Huang研究小组(表 1)[6, 7],其抑制活性IC50均采用荧光共振能量转移酶切割底物的荧光吸收强度测定。模拟过程中,随机选取50个分子组成训练集用于建立预测模型,其余分子组成验证集以对所建模型进行检验,分子的生物活性用pIC50(即-logIC50)表示。
 下载:
导出CSV
下载:
导出CSV
 |
||||||
| compd | xanthene core | R1 | R2 | IC50(nmol/L)(observed) | pIC50(observed) | pIC50(pridicted) |
| 1 | all-H | pyrimidin-5-yl | OCH2i-Pr | 8.00 | 8.10 | 8.33 |
| 2 | all-H | pyrimidin-5-yl | OCH2t-Bu | 2.00 | 8.70 | 8.93 |
| 3 | all-H | pyrimidin-5-yl |  |
3.10 | 8.51 | 8.52 |
| 4 | all-H | pyrimidin-5-yl | (CH2)2t-Bu | 2.00 | 8.70 | 8.71 |
| 5 | all-H | pyrimidin-5-yl |  |
7.00 | 8.15 | 8.11 |
| 6* | all-H | pyrimidin-5-yl |  |
4.00 | 8.40 | 8.32 |
| 7 | all-H | pyrimidin-5-yl |  |
6.00 | 8.22 | 8.25 |
| 8 | all-H | pyrimidin-5-yl |  |
0.17×102 | 7.77 | 7.72 |
| 9* | all-H | pyrimidin-5-yl |  |
0.11×102 | 7.96 | 8.13 |
| 10 | all-H | pyrimidin-5-yl |  |
7.00 | 8.15 | 8.40 |
| 11* | all-H | pyrimidin-5-yl |  |
7.00 | 8.15 | 7.89 |
| 12 | all-H | pyrimidin-5-yl |  |
4.00 | 8.40 | 8.27 |
| 13* | all-H | pyrimidin-5-yl |  |
8.00 | 8.10 | 7.85 |
| 14 | all-H | pyrimidin-5-yl |  |
9.00 | 8.05 | 7.94 |
| 15 | all-H | pyrimidin-5-yl |  |
7.00 | 8.15 | 8.02 |
| 16* | all-H | pyridine-2-yl | OCH2t-Bu | 0.23×102 | 7.64 | 7.77 |
| 17 | all-H | pyridine-4-yl | OCH2t-Bu | 3.23×102 | 6.49 | 6.65 |
| 18 | all-H | pyridine-3-yl | OCH2t-Bu | 4.00 | 8.40 | 8.41 |
| 19* | all-H | 4-F-pyridine-3-yl | OCH2t-Bu | 6.00 | 8.22 | 8.12 |
| 20 | all-H | 5-CN-pyridine-3-yl | OCH2t-Bu | 3.00 | 8.52 | 8.40 |
| 21 | all-H | 5-F-pyridine-3-yl | OCH2t-Bu | 4.00 | 8.40 | 8.23 |
| 22 | all-H | 5-OMe-pyridine-3-yl | OCH2t-Bu | 4.00 | 8.40 | 8.28 |
| 23 | all-H | 2-F-pyridine-3-yl | OCH2t-Bu | 0.80 | 9.10 | 8.83 |
| 24 | all-H | 2-F-6-F-pyridine-3-yl | OCH2t-Bu | 0.50 | 9.30 | 9.05 |
| 25 | all-H | pyridine-3-yl |  |
7.00 | 8.15 | 7.98 |
| 26 | 3-F | pyridine-3-yl |  |
4.00 | 8.40 | 8.08 |
| 27* | 4-F | pyridine-3-yl |  |
1.00 | 9.00 | 8.12 |
| 28 | 5-F | pyridine-3-yl |  |
0.71×102 | 7.15 | 7.81 |
| 29 | 4-F | 2-F-pyridine-3-yl |  |
0.40 | 9.40 | 9.26 |
| 30 | 4-F | 2-F-pyridine-3-yl |  |
9.00 | 8.05 | 8.13 |
| 31 | 4-F | 2-F-pyridine-3-yl |  |
3.00 | 8.52 | 8.63 |
| 32* | 4-F | 2-F-pyridine-3-yl |  |
4.00 | 8.40 | 8.59 |
| 33 | 4-F | 2-F-pyridine-3-yl |  |
5.00 | 8.30 | 8.38 |
| 34 | 4-F | 2-F-pyridine-3-yl |  |
7.00 | 8.15 | 8.38 |
| 35* | 4-F | 2-F-pyridine-3-yl |  |
3.00 | 8.52 | 7.72 |
| 36 | 4-F | 2-F-pyridine-3-yl |  |
0.12×102 | 7.92 | 7.91 |
| 37 | all-H | phenyl | OMe | 1.18×103 | 5.93 | 5.91 |
| 38 | all-H | 2-Me-phenyl | OMe | 7.02×103 | 5.15 | 5.08 |
| 39 | all-H | 3-Me-phenyl | OMe | 1.67×103 | 5.78 | 6.19 |
| 40 | all-H | 4-Me-phenyl | OMe | 1.78×103 | 5.75 | 5.50 |
| 41* | all-H | 2-Cl-phenyl | OMe | 4.88×103 | 5.31 | 6.18 |
| 42 | all-H | 3-Cl-phenyl | OMe | 0.13×103 | 6.89 | 6.38 |
| 43 | all-H | 4-Cl-phenyl | OMe | 2.02×103 | 5.69 | 5.67 |
| 44 | all-H | 4-CF3O-phenyl | OMe | 5.06×103 | 5.30 | 5.32 |
| 45* | all-H | 3-CF3O-phenyl | OMe | 2.76×103 | 5.56 | 6.07 |
| 46 | all-H | 2-F-3-Cl-phenyl | OMe | 1.17×103 | 5.93 | 6.17 |
| 47 | all-H | 2-F-4-Cl-phenyl | OMe | 1.56×103 | 5.81 | 5.69 |
| 48* | all-H | 2-F-5-Cl-phenyl | OMe | 0.28×103 | 6.55 | 6.38 |
| 49* | all-H | 2-F-3-MeO-phenyl | OMe | 0.90×103 | 6.05 | 5.93 |
| 50 | all-H | 2-F-5-MeO-phenyl | OMe | 0.74×103 | 6.13 | 6.04 |
| 51 | all-H | 2-F-4-Me-5-Cl-phenyl | OMe | 3.21×103 | 5.49 | 5.83 |
| 52 | all-H | 2-F-pyridin-3-yl | OMe | 0.80×103 | 6.10 | 6.50 |
| 53 | all-H | 4-F-pyridin-3-yl | OMe | 0.90×103 | 6.05 | 6.01 |
| 54 | all-H | 2-Cl-pyridin-4-yl | OMe | 2.11×103 | 5.68 | 5.75 |
| 55 | all-H | pyridin-4-yl | OMe | 3.90×103 | 5.41 | 5.17 |
| 57 | all-H | (S)-pyrimidin-5-yl | OMe | 1.07×102 | 6.97 | 6.81 |
| 58 | all-H | (R)-pyrimidin-5-yl | OMe | 1.04×104 | 4.98 | 4.94 |
| 59 | all-H | pyrimidin-5-yl | OPr | 0.16×102 | 7.80 | 7.73 |
| 60 | all-H | pyrimidin-5-yl | OBu | 8.00 | 8.10 | 8.17 |
 |
||||||
| compd | Ar | R | IC50(nmol/L)(observed) | pIC50(observed) | pIC50(pridicted) | |
| 56* | (R)-2-F-pyridin-3-yl | CHF2 | 1.21×103 | 5.92 | 5.11 | |
| 61* | 5-Cl-2-F-phenyl | Me | 1.95×103 | 5.71 | 5.90 | |
| 62 | 3-Cl-phenyl | Me | 2.06×103 | 5.69 | 5.77 | |
| 63 | pyridin-3-yl | Me | 7.46×103 | 5.13 | 5.08 | |
| 64 | pyrimidin-5-yl | Me | 1.14×104 | 4.94 | 5.01 | |
| 65* | 6-F-pyridin-3-yl | Me | 8.20×103 | 5.09 | 5.07 | |
| 66 | 2-F-pyridin-3-yl | Me | 2.85×103 | 5.55 | 5.49 | |
| 注:*表示验证集分子 | ||||||
预测模型的建立采用Tripos公司Sybyl 6.9程序包完成,软件中的各项参数除特别说明外均采用缺省值[8]。分子活性构象的确定是模型构建成功与否的关键因素。首先在Sketch molecule下确定分子的三维结构,并进行Minimize操作,原子电荷的计算采用Gasteiger-Huckel方法,以采用与距离相关介电函数的Tripos分子力场进行能量最小化,能量收敛梯度为0.05kcal·mol-1·Å-1。最终以最低能量对应的构象为最佳活性构象。分子叠合对模型具有重要影响,实验选取化合物中的呫吨部分(图式 1中的粗体部分)作为共同骨架结构,以最强抑制活性化合物29为模板分子进行叠合。最后运用CoMSIA法建立模型。
CoMSIA法认为,当一系列结构相似的化合物以相同的作用方式作用于同一受体时,化合物的生物活性就取决于每个化合物周围分子场能量势函数的分布。研究这些化合物周围的分子场分布,可以定量获得大量的分子场信息,以此作为自变量参与对药物生物活性的回归分析,从而建立预测分子生物活性的模型。CoMSIA法包括5种分子场,分别是立体场(steric,S)、静电场(electrostatic,E)、疏水场(hydrophobic,H)、氢键受体场(hydrogen-bond acceptor,A)以及氢键给体场(hydrogen-bond donor,D)[8]。该方法采用与距离相关的高斯函数作为分子场的能量函数,以sp3杂化的C+探针离子来计算各分子场的相似性指数[9]。分子场信息收集完成后,以偏最小二乘回归(partial least square,PLS)方法对收集的数据进行降维处理。过程中采用留一法(leave-one-out,LOO)进行交叉验证,并进一步回归分析得到分子场特征与分子活性之间的定量函数关系,最终拟合得到模型。
模型的检验分为内部检验和外部检验。采用非交叉验证方法(non-cross validation)对方程进行拟合程度的内部检验,得到主要评价指标非交叉验证相关系数Rncv2、标准偏差SEE[8]。外部检验通常运用样本外检验集分子进行,以外部验证相关系数Rpre2为评价指标。
最后,运用Stdev*Coeff法得到各分子场的三维等势面图,直观反映其对化合物活性的影响[8]。
分子对接可将药物分子配体与蛋白质受体通过空间匹配和能量匹配相互识别形成复合物,并预测该复合物的结构。分子对接把配体和受体的结合看作是一个能量释放的过程,结合能越低越稳定,因此,对接程序通常根据自由能数值的变化来评价配体的优劣。本实验采用的GOLD 5.1程序包对66个化合物进行分子对接,该程序采用遗传算法,其评分函数考虑了蛋白质与配体之间的氢键和范德华相互作用,以及配体的分子内氢键的贡献[10]。实验中的BACE1晶体结构(PDB代码:4RCF)来源于蛋白质数据库Protein Data Bank (www.rcsb.org/pdb),分辨率为1.78Å。对接过程中利用该晶体复合物中原配体所在的位置为参考,删除外源性配体,保留水分子,确定有效的受体-配体结合空腔,以遗传算法对配体进行构象优化,并对配体-受体之间的结合进行打分,每个配体分子保存打分排序最高的20个构象[8]。
通过LOO法对训练集分子5种分子场的所有组合进行回归分析,发现以S、H、A场建立的模型最佳(表 2),其交叉验证相关系数Q2高达0.86,最佳主成分数OPN为9。模型的内部检验结果也较理想,非交叉验证相关系数Rncv2为0.97,表明模型的内部预测能力较强。H和A场对模型的贡献率较大,均在40%以上,表明疏水基团和氢键受体基团对化合物的抑制活性具有较强的积极影响,立体场的影响较小。
下载:
导出CSV
| PLS | Q2 | Rncv2 | OPN | Rpre2 | SEE | Contribution | ||
| S | H | A | ||||||
| CoMSIA | 0.86 | 0.97 | 9 | 0.88 | 0.23 | 0.12 | 0.41 | 0.47 |
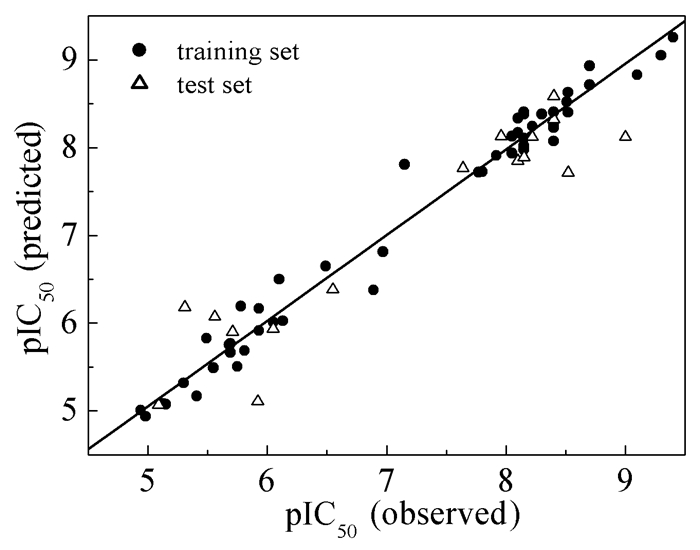
运用验证集分子统计分析得到的外部验证相关系数Rpre2为0.88,说明模型具有较强的外部预测能力和显著的统计意义,能够用于具有类似骨架结构BACE1抑制剂的活性预测。运用该模型对66个氨基噁唑啉呫吨化合物进行预测,得到的pIC50值见表 1。此外,从66个分子抑制活性的实验值与预测值的线性相关回归曲线(图 1)可以看出,所有数据点都集中分布在回归线两侧,表明基于训练集分子建立的CoMSIA模型相关性较好,稳定性和预测能力较强。
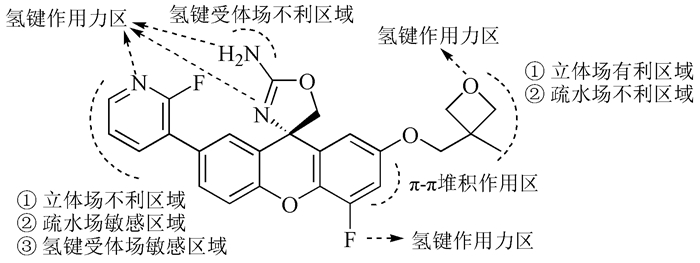
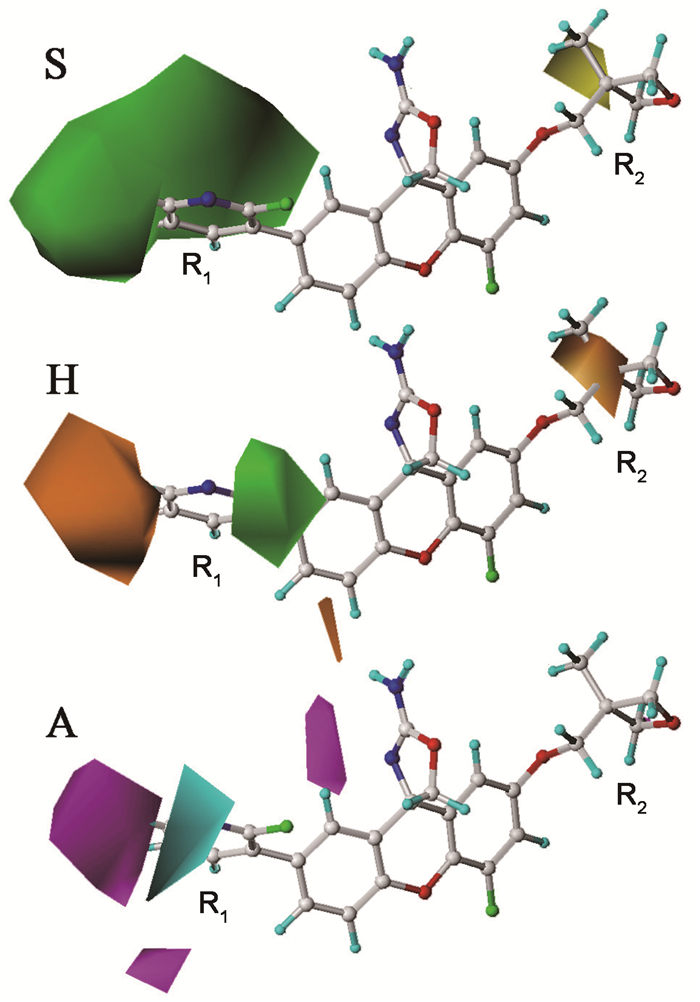
基于PLS分析的CoMSIA模型建立后,可以通过三维等势面图将各分子场对抑制剂分子生物活性的影响直观反映出来(图 2)。不同颜色的等值线图反映了不同性质的分子场影响,表示在相应分子场影响下,该区域基团有利于或不利于化合物抑制活性的增加,以此得到影响生物活性的分子结构特征信息,指导化合物的结构改造。从图 2中可以看出,三个分子场对氨基噁唑啉呫吨类抑制剂的影响主要集中在R1和R2基团上。
S:立体场;H:疏水场;A:氢键受体场
立体场S的三维等势面图中,黄色等值线表示在立体场影响下,该区域基团有利于抑制剂活性的增加,绿色等值线反之。R1取代基被一块大体积的绿色不利云团包围着,表明该区域不利于结合大体积基团,例如化合物44、45、51的R1取代基均为大体积基团,与结合小体积嘧啶基的57相比,分子活性明显偏低。立体场的黄色有利云团出现在R2取代基附近,意味着该区域链接大体积基团有利于分子抑制活性的提高。例如化合物57、59和60,它们R2取代基的体积依次增大,抑制活性也越来越强。此外,雄性幼大鼠的药效学实验结果显示,化合物60容易通过血脑屏障,能够显著降低中枢神经系统中的Aβ水平。当以100mg/kg的剂量单次口服给药化合物60时,4h后测得脑脊液中Aβ水平降低81%,脑中Aβ水平降低63%[6]。表明该系列化合物可能具有较强的CNS渗透性,明显的类药特性,值得进一步挖掘其结构特征以及与受体的作用模式。
疏水场H的绿色有利等值线云团出现在R1取代基附近,但R1取代基同时还被一块橙色不利云团包围着,说明该位置属于疏水场的敏感区域。另一块橙色不利等值线云团出现在R2取代基上,将氧杂环丁烷与甲基相连的碳原子紧紧包裹住,说明该位置结合疏水基团不利于分子生物活性的增强。例如,结合亲水基团的化合物11的抑制活性显著高于结合疏水基团的化合物57(表 3),与该实验得出的疏水场结论相一致。
下载:
导出CSV
 |
|||
| compd | R1 | R2 | pIC50 |
| 11 | pyrimidin-5-yl | OCH2C(CH3)2OH | 8.15 |
| 44 | 4-CF3O-phenyl | OMe | 5.30 |
| 45 | 3-CF3O-phenyl | OMe | 5.56 |
| 51 | 2-F-4-Me-5-Cl-phenyl | OMe | 5.49 |
| 57 | (S)-pyrimidin-5-yl | OMe | 6.97 |
| 59 | pyrimidin-5-yl | OPr | 7.80 |
| 60 | pyrimidin-5-yl | OBu | 8.10 |
氢键受体场A的蓝色有利等值线云团出现在R1取代基上方,同时该区域也出现了两块紫色不利云团,说明R1取代基是氢键受体场影响的敏感区域,此处结合包含氢键受体和供体的基团都有可能与蛋白质受体产生氢键作用力,使化合物的生物活性得到增强。有趣的是,本实验66个氨基噁唑啉呫吨化合物的R1基团均只含有氢键受体基团,未结合氢键供体基团,根据本实验结果,该类抑制剂在以后的结构改造中,可以尝试在该位置接入氢键供体基团来改善分子的抑制活性。另外,在氨基噁唑啉基团附近也出现了一块紫色不利云团,说明该区域结合包含氢键受体的基团不利于分子生物活性的改善。可以推测,该区域的氨基作为氢键供体基团很可能会与蛋白质受体产生氢键作用力。
分子对接通过优化和确定药物配体与蛋白质受体的最佳结合构象,能够预测它们之间的结合模式和特异性相互作用,在药物的结构改造中发挥着重要作用。本实验以最大活性分子29为模板化合物对该类抑制剂的对接结果进行讨论(图 3)。对接结果中显示的关键氨基酸与原文献晶体复合物(PDB:4RCF)中的对应信息完全一致[7],表明本实验准确定位了结合空腔的位置。
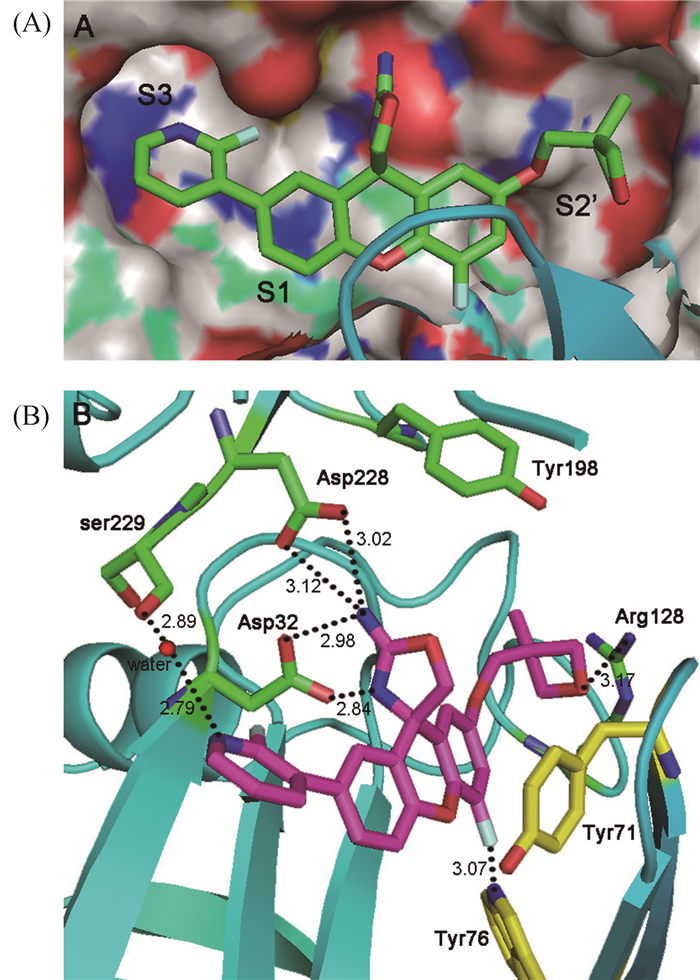
(A)化合物29与BACE1的结合空腔(PDB:4RCF);(B)化合物29与BACE1氨基酸残基的结合作用图
从图 3A可以看出,化合物29的氟代吡啶基团(R1取代基)占据了活性空腔的S3位点,该位点类似于一个圆形的洞穴,空间不大,因此大体积的R1取代基将不利于抑制剂活性的改善,这与前述的立体场结论一致。S3位点主要由疏水性氨基酸组成,仅包含少量的亲水性氨基酸,该位点本质上是疏水性的[11],并不利于与受体氨基酸产生氢键作用力。但是根据图 3B,亲水性氨基酸Ser229通过水分子与吡啶氮形成了较强的氢键作用力(—N…HO—,2.79Å;—OH…O—,2.89Å),对抑制剂生物活性的增强具有重要作用,因此R1取代基是疏水场和氢键受体场的敏感区域,表明了CoMSIA等值线结论的合理性和可靠性。
化合物29的氧杂环丁烷(R2取代基)占据了BACE1活性空腔的S2′位点(图 3(A)),该位点的空间较开阔,适宜结合大体积的基团,因此在图 2的等势面图中,该位置出现了立体场有利云团。该位点主要由亲水性氨基酸组成,本质上是亲水性的,因此占据该位点的亲水性配体基团将有可能与受体形成作用力,使分子的活性增强,正如图 3(B)中碱性氨基酸Arg128与氧杂环丁烷的受体氧形成的氢键作用力(—O…HN—,3.17 Å)。
化合物29的氨基噁唑啉作为功能基团与BACE1催化活性中心的Asp32和Asp228形成了四个氢键作用力,这与大多数的BACE1抑制剂的作用模式是一致的。另外,靠近R1取代基的呫吨苯环占据了分泌酶的S1位点;靠近R2取代基的呫吨苯环与氨基酸Tyr71形成了面-面π-π堆积作用,苯环4位上的氟原子作为氢键受体与氨基酸残基Tyr76产生了氢键作用力,这些结合特点对抑制剂生物活性的增强发挥了重要的作用。此外,幼鼠的药效学实验模型表明,在呫吨苯环4位上引入氟原子的化合物不仅会使其对BACE1的抑制活性增加,而且还能够导致幼鼠脑脊液和脑中Aβ水平的明显降低[7]。
水分子作为溶剂通常会在对接之前被去除[12]。然而,在溶剂化和去溶剂化过程中,水分子作为连接配体与蛋白质的桥梁,有助于配体-蛋白质之间的结合[13]。BACE1催化裂解APP的研究结果也表明,溶剂水分子有助于APP的Met和Asp间肽键的催化裂解[14],其重要性不可忽视。从本对接实验的结果(图 3(B))也可以看出,水分子作为氢键供体与R1取代基的吡啶氮形成了较强的作用力(—N…HO—,2.79Å),同时作为氢键供体又与氨基酸Ser229的羟基产生结合(—OH…O—,2.89Å),水分子在配体与受体蛋白的结合过程中起到了桥梁作用,对化合物的抑制作用具有重要影响。
本文的66个氨基噁唑啉呫吨类化合物属于小分子非肽类BACE1抑制剂,表现出了较强的抑制活性,发展潜力巨大。由立体场、疏水场和氢键受体场建立的构效关系模型预测能力强(Q2=0.86,Rncv2=0.97,Rpre2=0.88),结合分子对接结果,发现影响该类抑制剂生物活性的特征结构(图 4)为:(1)氨基噁唑啉取代基作为抑制剂的核心功能基团,与BACE1催化活性中心的两个氨基酸产生了较强的氢键结合,对分子生物活性的提高发挥了非常关键的作用;噁唑啉环上的氨基所在位置属于氢键受体场的不利区域;(2) R1取代基占据了分泌酶的S3活性位点,该区域适宜结合小体积且包含氢键受体的基团;(3) R2取代基占据了分泌酶的S2′活性位点,该位置结合大体积的亲水性基团将有利于化合物抑制活性的增强;(4)靠近R1取代基的呫吨苯环占据了分泌酶的S1活性位点,靠近R2取代基的呫吨苯环除了与受体形成了π-π堆积作用,其上的氟原子与受体产生了较强的氢键结合;(5)水分子对配体-受体之间的结合具有桥梁作用。以上信息是抑制剂具有强活性的关键因素,为后续新型BACE1抑制剂的结构改造和优化提供了重要参考。
M Citron. Nat. Rev. Drug Discov., 2010, 9:387~398. doi: 10.1038/nrd2896
D J Selkoe, J Hardy. EMBO Mol. Med., 2016, 8(6):595~608. doi: 10.15252/emmm.201606210
A K Ghosh, H L Osswald. Chem. Soc. Rev., 2014, 43:6765~6813. doi: 10.1039/C3CS60460H
C E H Moussa. Expert Opin. Inv. Drug., 2017, 26(10):1131~1136. doi: 10.1080/13543784.2017.1369527
R Yan. Transl. Neurodegener., 2016, 5:13. doi: 10.1186/s40035-016-0061-5
H Huang, D S La, A C Cheng et al. J. Med. Chem., 2012, 55:9156~9169. doi: 10.1021/jm300598e
O Epstein, M C Bryan, A C Cheng et al. J. Med. Chem., 2014, 57:9796~9810. doi: 10.1021/jm501266w
吴倩, 李先国, 李燕等.化学通报, 2016, 79(6):509~515. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20151018001&flag=1
吴倩, 高庆平, 王素青等.中国生物化学与分子生物学报, 2017, 33(11):1175~1181. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=SWHZ201711013&dbname=CJFD&dbcode=CJFQ
M L Verdonk, J C Cole, M J Hartshorn et al. Proteins, 2003, 52:609~623. doi: 10.1002/prot.10465
L Hong, G Koelsch, X Lin et al. Science, 2000, 290:150~153. doi: 10.1126/science.290.5489.150
D Van Der Spoel, E Lindahl, B Hess et al. J. Comput. Chem., 2005, 26:1701~1718. doi: 10.1002/(ISSN)1096-987X
E Aghaee, J B Ghasemi, F Manouchehri et al. J. Mol. Model., 2014, 20:1~13.
J Yuan, S Venkatraman, Y Zheng et al. J. Med. Chem., 2013, 56:4156~4180. doi: 10.1021/jm301659n

图式 1 包含共同骨架结构(粗体)的化合物29
Scheme 1 Molecular 29 containing the common substructure (bold)

图 1 抑制剂生物活性实验值与预测值的线性相关图
Figure 1 Plots of the observed versus the predicted pIC50 values

图 2 化合物29的CoMSIA三维等势面图
Figure 2 CoMSIA contour maps superimposed on 29
S:立体场;H:疏水场;A:氢键受体场

图 3 分子对接图
Figure 3 The docking results
(A)化合物29与BACE1的结合空腔(PDB:4RCF);(B)化合物29与BACE1氨基酸残基的结合作用图

图 4 氨基噁唑啉呫吨类BACE1抑制剂的特征结构图
Figure 4 The structural features sketch of aminooxazoline xanthenes as BACE1 inhibitors
表 1 氨基噁唑啉呫吨类BACE1抑制剂的结构和生物活性
Table 1. Structure and biological activity of aminooxazoline xanthene BACE1 inhibitors
|
||||||
| compd | xanthene core | R1 | R2 | IC50(nmol/L)(observed) | pIC50(observed) | pIC50(pridicted) |
| 1 | all-H | pyrimidin-5-yl | OCH2i-Pr | 8.00 | 8.10 | 8.33 |
| 2 | all-H | pyrimidin-5-yl | OCH2t-Bu | 2.00 | 8.70 | 8.93 |
| 3 | all-H | pyrimidin-5-yl | |
3.10 | 8.51 | 8.52 |
| 4 | all-H | pyrimidin-5-yl | (CH2)2t-Bu | 2.00 | 8.70 | 8.71 |
| 5 | all-H | pyrimidin-5-yl | |
7.00 | 8.15 | 8.11 |
| 6* | all-H | pyrimidin-5-yl | |
4.00 | 8.40 | 8.32 |
| 7 | all-H | pyrimidin-5-yl | |
6.00 | 8.22 | 8.25 |
| 8 | all-H | pyrimidin-5-yl | |
0.17×102 | 7.77 | 7.72 |
| 9* | all-H | pyrimidin-5-yl | |
0.11×102 | 7.96 | 8.13 |
| 10 | all-H | pyrimidin-5-yl | |
7.00 | 8.15 | 8.40 |
| 11* | all-H | pyrimidin-5-yl | |
7.00 | 8.15 | 7.89 |
| 12 | all-H | pyrimidin-5-yl | |
4.00 | 8.40 | 8.27 |
| 13* | all-H | pyrimidin-5-yl | |
8.00 | 8.10 | 7.85 |
| 14 | all-H | pyrimidin-5-yl | |
9.00 | 8.05 | 7.94 |
| 15 | all-H | pyrimidin-5-yl | |
7.00 | 8.15 | 8.02 |
| 16* | all-H | pyridine-2-yl | OCH2t-Bu | 0.23×102 | 7.64 | 7.77 |
| 17 | all-H | pyridine-4-yl | OCH2t-Bu | 3.23×102 | 6.49 | 6.65 |
| 18 | all-H | pyridine-3-yl | OCH2t-Bu | 4.00 | 8.40 | 8.41 |
| 19* | all-H | 4-F-pyridine-3-yl | OCH2t-Bu | 6.00 | 8.22 | 8.12 |
| 20 | all-H | 5-CN-pyridine-3-yl | OCH2t-Bu | 3.00 | 8.52 | 8.40 |
| 21 | all-H | 5-F-pyridine-3-yl | OCH2t-Bu | 4.00 | 8.40 | 8.23 |
| 22 | all-H | 5-OMe-pyridine-3-yl | OCH2t-Bu | 4.00 | 8.40 | 8.28 |
| 23 | all-H | 2-F-pyridine-3-yl | OCH2t-Bu | 0.80 | 9.10 | 8.83 |
| 24 | all-H | 2-F-6-F-pyridine-3-yl | OCH2t-Bu | 0.50 | 9.30 | 9.05 |
| 25 | all-H | pyridine-3-yl | |
7.00 | 8.15 | 7.98 |
| 26 | 3-F | pyridine-3-yl | |
4.00 | 8.40 | 8.08 |
| 27* | 4-F | pyridine-3-yl | |
1.00 | 9.00 | 8.12 |
| 28 | 5-F | pyridine-3-yl | |
0.71×102 | 7.15 | 7.81 |
| 29 | 4-F | 2-F-pyridine-3-yl | |
0.40 | 9.40 | 9.26 |
| 30 | 4-F | 2-F-pyridine-3-yl | |
9.00 | 8.05 | 8.13 |
| 31 | 4-F | 2-F-pyridine-3-yl | |
3.00 | 8.52 | 8.63 |
| 32* | 4-F | 2-F-pyridine-3-yl | |
4.00 | 8.40 | 8.59 |
| 33 | 4-F | 2-F-pyridine-3-yl | |
5.00 | 8.30 | 8.38 |
| 34 | 4-F | 2-F-pyridine-3-yl | |
7.00 | 8.15 | 8.38 |
| 35* | 4-F | 2-F-pyridine-3-yl | |
3.00 | 8.52 | 7.72 |
| 36 | 4-F | 2-F-pyridine-3-yl | |
0.12×102 | 7.92 | 7.91 |
| 37 | all-H | phenyl | OMe | 1.18×103 | 5.93 | 5.91 |
| 38 | all-H | 2-Me-phenyl | OMe | 7.02×103 | 5.15 | 5.08 |
| 39 | all-H | 3-Me-phenyl | OMe | 1.67×103 | 5.78 | 6.19 |
| 40 | all-H | 4-Me-phenyl | OMe | 1.78×103 | 5.75 | 5.50 |
| 41* | all-H | 2-Cl-phenyl | OMe | 4.88×103 | 5.31 | 6.18 |
| 42 | all-H | 3-Cl-phenyl | OMe | 0.13×103 | 6.89 | 6.38 |
| 43 | all-H | 4-Cl-phenyl | OMe | 2.02×103 | 5.69 | 5.67 |
| 44 | all-H | 4-CF3O-phenyl | OMe | 5.06×103 | 5.30 | 5.32 |
| 45* | all-H | 3-CF3O-phenyl | OMe | 2.76×103 | 5.56 | 6.07 |
| 46 | all-H | 2-F-3-Cl-phenyl | OMe | 1.17×103 | 5.93 | 6.17 |
| 47 | all-H | 2-F-4-Cl-phenyl | OMe | 1.56×103 | 5.81 | 5.69 |
| 48* | all-H | 2-F-5-Cl-phenyl | OMe | 0.28×103 | 6.55 | 6.38 |
| 49* | all-H | 2-F-3-MeO-phenyl | OMe | 0.90×103 | 6.05 | 5.93 |
| 50 | all-H | 2-F-5-MeO-phenyl | OMe | 0.74×103 | 6.13 | 6.04 |
| 51 | all-H | 2-F-4-Me-5-Cl-phenyl | OMe | 3.21×103 | 5.49 | 5.83 |
| 52 | all-H | 2-F-pyridin-3-yl | OMe | 0.80×103 | 6.10 | 6.50 |
| 53 | all-H | 4-F-pyridin-3-yl | OMe | 0.90×103 | 6.05 | 6.01 |
| 54 | all-H | 2-Cl-pyridin-4-yl | OMe | 2.11×103 | 5.68 | 5.75 |
| 55 | all-H | pyridin-4-yl | OMe | 3.90×103 | 5.41 | 5.17 |
| 57 | all-H | (S)-pyrimidin-5-yl | OMe | 1.07×102 | 6.97 | 6.81 |
| 58 | all-H | (R)-pyrimidin-5-yl | OMe | 1.04×104 | 4.98 | 4.94 |
| 59 | all-H | pyrimidin-5-yl | OPr | 0.16×102 | 7.80 | 7.73 |
| 60 | all-H | pyrimidin-5-yl | OBu | 8.00 | 8.10 | 8.17 |
|
||||||
| compd | Ar | R | IC50(nmol/L)(observed) | pIC50(observed) | pIC50(pridicted) | |
| 56* | (R)-2-F-pyridin-3-yl | CHF2 | 1.21×103 | 5.92 | 5.11 | |
| 61* | 5-Cl-2-F-phenyl | Me | 1.95×103 | 5.71 | 5.90 | |
| 62 | 3-Cl-phenyl | Me | 2.06×103 | 5.69 | 5.77 | |
| 63 | pyridin-3-yl | Me | 7.46×103 | 5.13 | 5.08 | |
| 64 | pyrimidin-5-yl | Me | 1.14×104 | 4.94 | 5.01 | |
| 65* | 6-F-pyridin-3-yl | Me | 8.20×103 | 5.09 | 5.07 | |
| 66 | 2-F-pyridin-3-yl | Me | 2.85×103 | 5.55 | 5.49 | |
| 注:*表示验证集分子 | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 CoMSIA模型的PLS分析结果
Table 2. PLS statistics of CoMSIA model
| PLS | Q2 | Rncv2 | OPN | Rpre2 | SEE | Contribution | ||
| S | H | A | ||||||
| CoMSIA | 0.86 | 0.97 | 9 | 0.88 | 0.23 | 0.12 | 0.41 | 0.47 |
下载: 导出CSV
表 3 氨基噁唑啉呫吨类BACE1抑制剂结构与活性数据
Table 3. Structure-activity relationship data related to aminooxazoline xanthenes as BACE1 inhibitors.
|
|||
| compd | R1 | R2 | pIC50 |
| 11 | pyrimidin-5-yl | OCH2C(CH3)2OH | 8.15 |
| 44 | 4-CF3O-phenyl | OMe | 5.30 |
| 45 | 3-CF3O-phenyl | OMe | 5.56 |
| 51 | 2-F-4-Me-5-Cl-phenyl | OMe | 5.49 |
| 57 | (S)-pyrimidin-5-yl | OMe | 6.97 |
| 59 | pyrimidin-5-yl | OPr | 7.80 |
| 60 | pyrimidin-5-yl | OBu | 8.10 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们