图 1.
一些具有代表性的Daphnilactone B类的虎皮楠生物碱
Figure 1.
Some representative daphnilactone B-type alkaloids

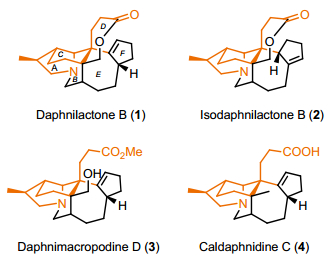
虎皮楠生物碱是生物碱家族中的一大类, 距今为止已被分离得到近300多个成员[1]. 1972年, Sasaki和Hirata小组[2]从虎皮楠科植物豆腐树的果实中提取得到一种具有六环骨架的虎皮楠生物碱Daphnilactone B (1), 并用X射线单晶衍射证明了其结构.该生物碱具有7个手性中心, 其中包括6个连续的手性中心, 空间上高度拥挤的全碳双季碳结构以及一个七元内酯环, 这是Daphnilactone B类型虎皮楠生物碱的第一个成员.之后科学家们又陆续从虎皮楠科植物中提取得到其它Daphnilactone B类型的生物碱.比如Yamamura[3], 郝小江[4], 岳建民[5]等先后分离得到了Isodaphnilactone B (2), Daphnimacropodine D (3), Caldaphnidine C (4)等(图 1).
近年来, 虎皮楠生物碱成为了天然产物全合成领域科学家们的研究热点[1c, 6]. Heathcock[7]、Carreira[8]、Smith[9]、李昂[10]、Hanessian[11]、Fukuyama[12]、翟宏斌[13]、Dixon[14]等小组均有非常精彩的全合成工作报道.但迄今为止, 仍没有课题组完成对Daphnilactone B这一类生物碱的全合成工作. 2006~2009年, Denmark小组[15, 16]利用不同的策略分别构建了这类生物碱骨架的ABC三环骨架. 2011年, Coldham小组[17]采用新策略构建Daphnilactone B的ABC环系.
本文采用了Diels-Alder (D-A)反应以及Conia-ene反应作为本路线的关键步骤, 经历七步反应高效构建了Daphnilactone B的AC环, 为此类生物碱的全合成工作奠定基础.
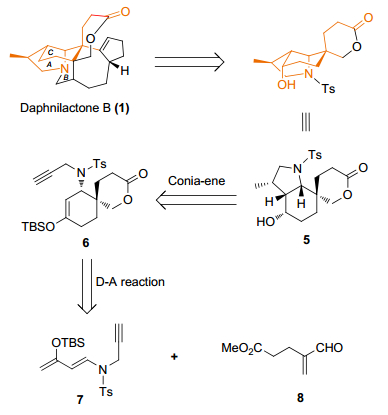
我们以D-A反应和Conia-ene反应为核心策略, 设计了合成虎皮楠生物碱Daphnilactone B的路线.对目标产物进行逆合成分析可知(Scheme 1), 我们使用Conia-ene反应[10, 18]可构建目标产物的A环, 左侧的甲基通过氢化双键来得到.再设计通过分子间的Diels-Alder反应来构建目标产物的C环, 而D-A反应的Rawal二烯[19]的类似物7可通过aza-Michael加成反应来得到, 亲双烯体8可根据文献已报道的方法合成[20].
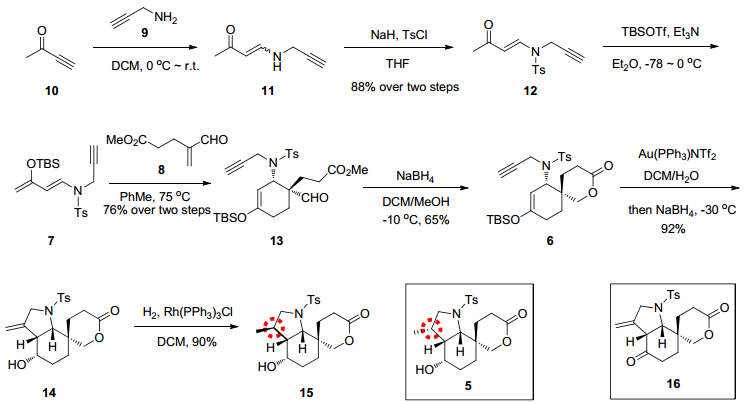
根据以上逆合成分析, 合成路线如Scheme 2所示.我们首先合成Rawal二烯的类似物7, 以化合物9和10作为原料出发, 经aza-Michael加成反应得到中间体11 (Z:E=10:1), 无需后处理纯化直接将反应溶剂蒸干即可作为原料进行下一步反应.由于裸露的氨基可能会给后续的反应带来影响, 所以将氨基进行保护.综合考量之下, 最终选择对甲苯磺酰基(Ts)作为保护基. 0 ℃下, 用氢化钠作为碱, 在四氢呋喃中使用对甲苯磺酰氯(TsCl)将化合物11中的氮原子保护, 以90%的产率得到化合物12, 且几乎都是反式结构.最终使用叔丁基二甲硅基三氟甲磺酸酯(TBSOTf)和三乙胺(Et3N)将化合物12中的甲基酮结构转化为烯醇硅醚.最初使用二氯甲烷作为硅醚化反应的溶剂, 产率只有50%.经过不断尝试, 最终发现在乙醚作为该反应的溶剂时, 以接近定量的收率得到双烯体化合物7.
在成功制备双烯体化合物7后, 尝试使用Diels-Alder反应构建目标化合物C环.我们将双烯体化合物7和亲双烯体化合物8在甲苯中加热, 经过温度条件筛选, 最终发现在75 ℃下化合物7和8在甲苯中可成功发生D-A反应, 以76%的产率得到化合物13 (dr>10:1).
我们尝试用Conia-ene反应来构建目标化合物A环, 该反应在李昂小组的多个对于虎皮楠生物碱的精彩全合成工作当中均得到成功应用[10].但是在一系列的尝试之后, 都不能以较好的收率得到环化产物.可能是由于炔与金离子络合之后, 受到醛羰基的影响.
由此, 我们决定先将醛基还原以解决上述问题.在-10 ℃下, 以二氯甲烷/甲醇作为混合溶剂, 用硼氢化钠还原化合物13中的醛基, 生成伯醇后随即发生分子内的酯交换反应, 得到内酯化合物6.接下来, 以化合物6作为底物尝试通过金催化的Conia-ene反应来构建目标化合物A环[10].经过一系列条件筛选(如表 1)发现, 使用10 mol%的三苯基膦金(I)双(三氟甲磺酰基)亚胺盐(Au(PPh3)NTf2), 并以V(二氯甲烷):V(H2O)=10:1作为溶剂常温下即可快速得到环化产物16.但是化合物16不稳定, 直接将未经柱层析纯化的粗产品用硼氢化钠将酮羰基还原, 得到化合物14.但是经核磁图谱判断, 化合物14的羟基朝向平面内侧, 可能对于下一步氢化具有和预期相反的诱导作用.我们将化合物14在二氯甲烷作溶剂, 氢气氛围下用Wilkinson催化剂(Rh(PPh3)3Cl)催化[21].结果正如我们所预料的, 以90%的收率得到了甲基与最初预期构型构型相反的产物15 (dr>20:1).
 下载:
导出CSV
下载:
导出CSV
| 序号 | 催化剂 | 添加剂 | 溶剂 | 产率/% |
| 1 | Au(PPh3)Cl (2 mol%) | AgBF4 (2 mol%) | V(PhMe):V(MeOH)=10:1 | 44 |
| 2 | Au(PPh3)Cl (2 mol%) | AgBF4 (2 mol%) | V(DCM):V(H2O)=10:1 | 52 |
| 3 | Au(PPh3)NTf2 (2 mol%) | — | V(PhMe):V(MeOH)=10:1 | 62 |
| 4 | Au(PPh3)NTf2 (10 mol%) | — | V(PhMe):V(MeOH)=10:1 | 69 |
| 5 | Au(PPh3)NTf2 (10 mol%) | — | V(DCM):V(H2O)=10:1 | 95 |
| 6 | Au(PPh3)NTf2 (10 mol%) | — | V(丙酮):V(H2O)=10:1 | 57 |
| 7 | Au(PPh3)NTf2 (10 mol%) | — | V(丙酮):V(丙酸)=20:1 | 62 |
| 8 | Au(PPh3)NTf2 (10 mol%) | — | V(DCM):V(丙酸)=20:1 | 81 |
| 9 | Au(PPh3)NTf2 (10 mol%) | — | V(DCM):V(MeOH)=10:1 | 66 |
随后我们计划将化合物14中的羟基通过Mistunobu反应进行构型调整, 再进行诱导氢化.后续考虑将内酯环通过胺酯交换反应打开, 并对生成的伯醇进行氧化以及后续的修饰, 从而达到引入第一个双季碳结构的目的, 后续研究正在进行中.
通过以商业可得的原料合成了化合物14, 首先使用了非常高效的D-A反应构建了Daphnilactone的C环, 然后通过金催化的Conia-ene反应构建了A环, 并且为随后引入季碳结构做了很好的铺垫, 为部分Daphnilactone B类虎皮楠生物碱提供了一个较好的AC环合成策略.
无水无氧的实验均使用Schlenk技术在氩气保护下进行, 溶剂均经除水蒸馏处理.二氯甲烷(DCM)使用CaH2干燥除水后蒸馏, 四氢呋喃(THF)、甲苯、Et2O采用Na除水并用二苯甲酮作为指示剂, 三乙胺使用前用CaH2干燥后重蒸.所有使用的试剂均为分析级纯度.薄层硅胶板用紫外灯或者使用高锰酸钾显色剂来显色.柱层析所使用的硅胶粒度为200~300目.核磁共振氢谱、碳谱所使用的仪器为Bruker系列的400M/500M核磁共振仪.高分辨质谱数据由赛默飞世尔科技Q Exactive系列组合型四极杆Orbitrap质谱仪测定.
化合物8按照文献[20]的方法合成.
在氩气氛围下, 将乙炔基甲基酮(10) (15.0 g, 220 mmol, 17.2 mL)溶于70 mL无水二氯甲烷.将体系冷却至0 ℃, 将炔丙胺(9) (11.0 g, 200 mmol, 12.8 mL)缓缓加入上述体系中.在0 ℃下搅拌10 min, 缓慢升至室温.继续搅拌至原料反应完全后浓缩.将浓缩后的粗产品在氩气氛围下溶于600 mL无水四氢呋喃中, 冷却至-10 ℃.分批加入氢化钠(含量60%, 9.6 g, 240 mmol), 升温至0 ℃搅拌10 min, 将体系冷却至-20 ℃, 加入对甲苯磺酰氯(45.8 g, 240 mmol).在0 ℃下搅拌3 h,用饱和碳酸氢钠溶液小心淬灭, 加入乙酸乙酯萃取(120 mL×3), 合并有机相, 并用饱和食盐水100 mL洗涤无水硫酸钠干燥, 浓缩.将浓缩后得到的粗产品用柱层析纯化[V(石油醚):V(乙酸乙酯)=4:1]得白色固体12 (48.0 g, 两步收率为85%). m.p. 91~93 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.92 (d, J=14.2 Hz, 1H), 7.72 (d, J=8.4 Hz, 2H), 7.33 (d, J=8.1 Hz, 2H), 5.65 (d, J=14.2 Hz, 1H), 4.31 (d, J=2.5 Hz, 2H), 2.42 (s, 3H), 2.23 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 196.38, 145.22, 139.85, 134.85, 130.06, 127.46, 109.75, 74.81, 74.23, 35.31, 27.91, 21.64; HRMS-ESI calcd for C14H16NO3S [M+H+] 278.0845, found 278.0846.
在氩气氛围下, 将化合物12 (6.8 g, 24.5 mmol)溶于无水乙醚中.将上述体系冷却至-78 ℃, 加入三乙胺(10.3 mL, 73.5 mmol), 然后缓缓加入叔丁基二甲硅基三氟甲磺酸酯(8.46 mL, 36.7 mmol)后搅拌30 min, 再升温至0 ℃搅拌至原料全部转化.缓慢加入80 mL饱和碳酸氢钠水溶液, 有机相用饱和食盐水洗涤(100 mL×2), 无水硫酸钠干燥, 浓缩.
将浓缩得到的粗产品和化合物8 (6.97 g, 49 mmol)在氩气氛围下溶于81 mL无水甲苯中.将体系加热至75 ℃, 搅拌2 d.加入2 mL三乙胺, 将反应体系浓缩得粗产品.将粗产品用柱层析进行分离纯化[V(石油醚):V(乙酸乙酯):V(三乙胺)=1000:60:20~1000:100:25, 硅胶柱用含5%三乙胺的石油醚饱和], 得无色泡沫状产物13 (9.95 g, 两步收率为76%). 1H NMR (400 MHz, CDCl3) δ: 9.78 (s, 1H), 7.73 (d, J=8.2 Hz, 2H), 7.31 (d, J=8.0 Hz, 2H), 4.62 (d, J=5.0 Hz, 1H), 3.83 (s, 2H), 3.77 (d, J=5.1 Hz, 1H), 3.64 (s, 3H), 2.41 (s, 3H), 2.19~2.16 (m, 1H), 2.14~1.86 (m, 6H), 1.76~1.64 (m, 2H), 0.80 (s, 9H), -0.06 (s, 3H), -0.10 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 203.18, 173.05, 156.91, 144.13, 136.23, 129.88, 127.42, 97.20, 79.39, 72.80, 59.24, 51.86, 49.60, 34.38, 28.52, 26.79, 25.46, 25.30, 21.52, 20.23, 17.85, -4.59, -4.78; HRMS-ESI calcd for C27H40NO6SSi [M+H+] 534.2340, found 534.2338.
将化合物13 (3.70 g, 6.94 mmol)溶于33 mL二氯甲烷和甲醇[V(DCM):V(MeOH)=10:1]的混合溶剂中, 将体系冷却至-10 ℃, 分批加入硼氢化钠(0.13 g, 3.45 mmol).搅拌至原料全部消耗完毕.小心加入40 mL饱和碳酸氢钠溶液淬灭.将体系升至室温后继续搅拌15 min.加入2 mL三乙胺, 再加入二氯甲烷萃取(50 mL×4), 合并有机相并用饱和食盐水洗涤(50 mL)后用无水硫酸钠干燥, 浓缩.将浓缩所得粗产品进行柱层析分离纯化[V(石油醚):V(乙酸乙酯)=1:0~5:1~4:1, 硅胶柱用含5%三乙胺的石油醚饱和]得无色泡沫状产物6 (2.24 g, 产率为65%). 1H NMR (500 MHz, CDCl3) δ: 7.79~7.69 (m, 2H), 7.33 (d, J=8.3 Hz, 2H), 4.53 (d, J=12.0 Hz, 1H), 4.43~4.36 (m, 1H), 4.28 (d, J=5.5 Hz, 1H), 3.96~3.86 (m, 1H), 3.80~3.70 (m, 2H), 2.65 (ddd, J=16.3, 9.0, 6.9 Hz, 1H), 2.50~2.42 (m, 3H), 2.36~2.28 (m, 1H), 2.12~1.99 (m, 2H), 1.99~1.85 (m, 1H), 1.79 (tt, J=9.5, 4.9 Hz, 2H), 1.73~1.64 (m, 1H), 0.82 (s, 9H), -0.06 (s, 3H), -0.09 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 172.48, 157.64, 144.12, 136.17, 129.94, 127.11, 97.46, 80.20, 74.71, 72.89, 60.60, 60.37, 34.56, 34.25, 28.63, 27.16, 25.55, 25.42, 21.48, 17.86, -4.63, -4.83; HRMS-ESI calcd for C26H38NO5SSi [M+H+] 504.2234, found 504.2231.
在氩气氛围下, 将化合物6 (1.42 g, 2.82 mmol)溶于16 mL二氯甲烷和水[V(DCM):V(H2O)=10:1)]的混合溶剂中.加入三苯基膦金(I)双(三氟甲磺酰基)亚胺盐(209 mg, 0.282 mmol), 搅拌15 min.用10 mL饱和碳酸氢钠溶液和10 mL饱和食盐水淬灭, 加入二氯甲烷萃取(10 mL×3), 合并有机相并用饱和食盐水洗涤(10 mL)后干燥, 浓缩.在氩气氛围下, 将浓缩所得的粗产品溶于32 mL的四氢呋喃和甲醇(V:V=3:1)混合溶剂中, 并冷却至-30℃.分批加入硼氢化钠(53.3 mg, 1.41 mmol), 搅拌90 min, 加入8 mL甲醇.将反应体系浓缩, 加入二氯甲烷萃取(15 mL×4), 合并有机相并用饱和食盐水(15 mL)洗涤.用无水硫酸钠干燥后浓缩.将浓缩所得粗产品用柱层析进行分离纯化[V(石油醚):V(乙酸乙酯)=1:1~3:2]得无色泡沫状产物14 (1.02 g, 产率92%). 1H NMR (500 MHz, CDCl3) δ: 7.68 (d, J=8.1 Hz, 2H), 7.32 (d, J=8.0 Hz, 2H), 5.58 (d, J=2.2 Hz, 1H), 5.01 (d, J=2.2 Hz, 1H), 4.41 (dd, J=11.3, 2.3 Hz, 1H), 4.17 (dd, J=11.2, 1.4 Hz, 1H), 3.95 (ddd, J=12.7, 5.4, 2.6 Hz, 2H), 3.82~3.72 (m, 2H), 2.79~2.62 (m, 2H), 2.49 (dt, J=17.5, 7.3 Hz, 1H), 2.42 (s, 3H), 2.27 (d, J=7.9 Hz, 1H), 1.96 (d, J=14.5 Hz, 1H), 1.88~1.76 (m, 1H), 1.63 (dt, J=13.4, 4.0 Hz, 1H), 1.38 (dddd, J=12.7, 6.9, 5.4, 2.2 Hz, 1H), 1.09 (td, J=14.4, 3.6 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ: 172.21, 144.48, 143.19, 133.91, 130.22, 127.56, 109.57, 69.83, 69.76, 67.68, 54.35, 46.24, 37.56, 29.37, 27.52, 25.17, 21.61; HRMS-ESI calcd for C20H25N- O5SNa [M+Na+] 414.1346, found 414.1342.
将化合物14 (0.650 g, 1.65 mmol)和三(三苯基膦)氯化铑(I) (0.153 g, 0.165 mmol)在氮气氛围下溶于40 mL无水二氯甲烷中.将体系用氢气置换三次, 使体系在氢气氛围下(101 kPa), 搅拌过夜.将反应液浓缩后用柱层析分离纯化[V(石油醚):V(乙酸乙酯)=1:1]得无色油状液体5 (0.580 g, 产率为90%). 1H NMR (500 MHz, CDCl3) δ: 7.75~7.71 (m, 2H), 7.35 (d, J=8.0 Hz, 2H), 5.00 (dd, J=11.6, 1.6 Hz, 1H), 4.66 (d, J=11.8 Hz, 1H), 4.14 (dq, J=6.3, 2.8 Hz, 1H), 3.69 (d, J=8.2 Hz, 1H), 3.52 (dd, J=11.6, 8.1 Hz, 1H), 3.37 (t, J=11.5 Hz, 1H), 2.94~2.78 (m, 2H), 2.51~2.46 (m, 1H), 2.45 (s, 3H), 1.99 (td, J=7.9, 3.9 Hz, 1H), 1.91 (dtd, J=14.7, 7.0, 2.5 Hz, 1H), 1.81 (ddd, J=14.2, 6.4, 2.5 Hz, 1H), 1.72 (dddd, J=14.6, 11.4, 6.4, 2.5 Hz, 1H), 1.59 (dq, J=11.2, 7.4 Hz, 1H), 1.52~1.43 (m, 1H), 1.17 (ddd, J=14.0, 11.3, 7.0 Hz, 1H), 1.02 (d, J=6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 174.72, 143.95, 134.15, 129.92, 127.75, 70.01, 67.78, 64.55, 57.39, 44.84, 35.96, 34.30, 33.94, 29.23, 29.06, 28.07, 21.60, 11.90; HRMS-ESI calcd for C20H27N- O5SNa[M+Na+] 416.1502, found 416.1499.
辅助材料(Supporting Information) 包含本路线中经分离纯化得到的新化合物1H NMR, 13C NMR, 以及化合物15的二维核磁谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Kobayashi, J.; Kubota, T.Nat.Prod.Rep.2009, 26, 936.
(b) Li, Z.-Y.; Guo, Y.-W.Chin.J.Org.Chem.2007, 27, 565 (in Chinese).
(李震宇, 郭跃伟, 有机化学, 2007, 27, 565.) (c) Chattopadhyay, A.K.; Hanessian, S.Chem.Rev.2017, 117, 4104.
(d) Kang, B.; Jakubec, P.; Dixon, D.J.Nat.Prod.Rep.2014, 31, 550.
(e) Yang, S.-P.; Yue, J.-M.Acta Pharmacol.Sin.2012, 33, 1147.
Sasaki, K.; Hirata, Y.Tetrahedron Lett.1972, 13, 1891. doi: 10.1016/S0040-4039(01)84744-4
Yamamura, S.; Terada, Y.Chem.Lett.1976, 5, 1381. doi: 10.1246/cl.1976.1381
Kong, N.-C.; He, H.-P.; Wang, Y.-H.; Mu, S.-Z.; Di, Y.-T.; Hao, X.-J.J.Nat.Prod.2007, 70, 1348. doi: 10.1021/np0700220
Zhan, Z.-J.; Zhang, C.-R.; Yue, J.-M.Tetrahedron 2005, 61, 11038. doi: 10.1016/j.tet.2005.03.146
For synthetic studies published after Ref.[1c], see:
(a) Li, J.L.; Shi, H.W.; Wang, Q.; Chai, Y.H.; Yang, J.Org.Lett.2017, 19, 1497.
(b) Boissarie, P.; Bélanger, G.Org.Lett.2017, 19, 3739.
(c) Liu, Y.M.; Li, F.; Wang, Q.; Yang, J.Tetrahedron 2017, 73, 6381.
(d) Shao, H.; Bao, W.; Jing, Z.-R.; Wang, Y.-P.; Zhang, F.-M.; Wang, S.-H.; Tu, Y.-Q.Org.Lett.2017, 19, 4648.
(e) Lopez, A.M.; Ibrahim, A.A.; Rosenhauer, G.J.; Sirinimal, H.S.; Stockdill, J.L.Org.Lett.2018, 20, 2216.
(f) Sasano, Y.; Koyama, J.; Yoshikawa, K.; Kanoh, N.; Kwon, E.; Iwabuchi, Y.Org.Lett.2018, 20, 3053.
(g) Yamada, R.; Fukuyama, T.; Yokoshima, S.Org.Lett.2018, 20, 4504.
(h) Li, Y.; Dong, Q.; Xie, Q.; Tang, P.; Zhang, M.; Qin, Y.Org.Lett.2018, 20, 5053.
(i) Qiu, Y.; Zhong, J.; Du, S.; Gao, S.Chem.Commun.2018, 54, 5554.
(j) Kitabayashi, Y.; Fukuyama, T.; Yokoshima, S.Org.Biomol.Chem.2018, 16, 3556.
(k) Mo, X.-F.; Li, Y.-F.; Sun, M.-H.; Dong, Q.-Y.; Xie, Q.-X.; Tang, P.; Xue, F.; Qin, Y.Tetrahedron Lett.2018, 59, 1999.
(l) Cox, J.B.; Wood, J.L.Tetrahedron 2018, 74, 4539.
(a) Heathcock, C.H.; Davidsen, S.K; Mills, S.; Sanner, M.A.J.Am.Chem.Soc.1986, 108, 5650.
(b) Ruggeri, R.B.; Hansen, M.M.; Heathcock, C.H.J.Am.Chem.Soc.1988, 110, 8734.
(c) Ruggeri, R.B.; McClure, K.F.; Heathcock, C.H.J.Am.Chem.Soc.1989, 111, 1530.
(d) Ruggeri, R.B.; Heathcock, C.H.J.Org.Chem.1990, 55, 3714.
(e) Stafford, J.A.; Heathcock, C.H.J.Org.Chem.1990, 55, 5433.
(f) Piettre, S.; Heathcock, C.H.Science 1990, 248, 1532.
(g) Heathcock, C.H.; Stafford, J.A.; Clark, D.L.J.Org.Chem.1992, 57, 2575.
(h) Heathcock, C.H.Angew.Chem., Int.Ed.Engl.1992, 31, 665.
(i) Heathcock, C.H.; Joe, D.J.Org.Chem.1995, 60, 1131.
(j) Heathcock, C.H.; Kath, J.C.; Ruggeri, R.B.J.Org.Chem.1995, 60, 1120.
(k) Heathcock, C.H.Proc.Natl.Acad.Sci.U.S.A. 1996, 93, 14323.
(l) Wallace, G.A.; Heathcock, C.H.J.Org.Chem.2001, 66, 450.
Weiss, M.E.; Carreira, E.M.Angew.Chem., Int.Ed.2011, 50, 11501. doi: 10.1002/anie.v50.48
(a) Shvartsbart, A.; Smith, A.B.Ⅲ J.Am.Chem.Soc.2014, 136, 870.
(b) Shvartsbart, A.; Smith, A.B.Ⅲ J.Am.Chem.Soc.2015, 137, 3510.(a) Shvartsbart, A.; Smith, A.B.Ⅲ J.Am.Chem.Soc.2014, 136, 870.
(b) Shvartsbart, A.; Smith, A.B.Ⅲ J.Am.Chem.Soc.2015, 137, 3510.
(a) Lu, Z.; Li, Y.; Deng, J.; Li, A.Nat.Chem.2013, 5, 679.
(b) Xiong, X.; Li, Y.; Lu, Z.; Wan, M.; Deng, J.; Wu, S.; Shao, H.; Li, A.Chem.Commun.2014, 50, 5294.
(c) Li, J.; Zhang, W.; Zhang, F.; Chen, Y.; Li, A.J.Am.Chem.Soc.2017, 139, 14893.
(d) Chen, Y.; Zhang, W.; Ren, L.; Li, J.; Li, A.Angew.Chem., Int.Ed.2018, 57, 952.
(e) Zhang, W.; Ding, M.; Li, J.; Guo, Z.; Lu, M.; Chen, Y.; Liu, L.; Shen, Y.-H.; Li, A.J.Am.Chem.Soc.2018, 140, 4227.
For the total synthesis of a putative natural Daphniphyllum alkaloid isodaphlongamine H: Chattopadhyay, A.K.; Ly, V.L.; Jakkepally, S.; Berger, G.; Hanessian, S.Angew.Chem., Int.Ed.2016, 55, 2577.
Yamada, R.; Adachi, Y.; Yokoshima, S.; Fukuyama, T.Angew.Chem., Int.Ed.2016, 55, 6067. doi: 10.1002/anie.201601958
Chen, X.; Zhang, H.-J.; Yang, X.; Lv, H.; Shao, X.; Tao, C.; Wang, H.; Cheng, B.; Li, Y.; Guo, J.; Zhang, J.; Zhai, H.Angew.Chem., Int.Ed.2018, 57, 947. doi: 10.1002/anie.201709762
Shi, H.; Michaelides, I.N.; Darses, B.; Jakubec, P.; Nguyen, Q.N.N.; Paton, R.S.; Dixon, D.J.J.Am.Chem.Soc.2017, 139, 17755. doi: 10.1021/jacs.7b10956
Denmark, S.E.; Baiazitov, R.Y.J.Org.Chem.2006, 71, 593. doi: 10.1021/jo052001l
(a) Denmark, S.E.; Baiazitov, R.Y.; Nguyen, S.T.Tetrahedron 2009, 65, 6535.
(b) Denmark, S.E.; Nguyen, S.T.; Baiazitov, R.Y.Heterocycles 2008, 76, 143.
Coldham, I.; Burrell, A.J.M.; Guerrand, H.D.S.; Oram, N.Org.Lett.2011, 13, 1267. doi: 10.1021/ol102961x
(a) Staben, S.T.; Kennedy-Smith, J.J.; Huang, D.; Corkey, B.K.; Lalonde, R.L.; Toste, F.D.Angew.Chem., Int.Ed.2006, 45, 5991.
(b) Huwyler, N.; Carreira, E.M.Angew.Chem., Int.Ed.2012, 51, 13066. For a Conia-ene reaction catalyzed by Rh, see: :
(c) Long, R.; Huang, J.; Shao, W.; Liu, S.; Lan, Y.; Gong, J.; Yang, Z.Nat.Commun.2014, 5, 5707.
Kozmin, S.A.; Rawal, V.H.J.Org.Chem.1997, 62, 5252. doi: 10.1021/jo970438q
Terrasson, V.; van der Lee, A.; Marcia de Figueiredo, R.; Campagne, J.M.Chem.-Eur.J.2010, 16, 7875. doi: 10.1002/chem.201000334
Zhang, P.-P.; Yan, Z.M.; Li, Y.-H.; Gong, J.-X.; Yang, Z.J.Am.Chem.Soc.2017, 139, 13989. doi: 10.1021/jacs.7b07388

图 1 一些具有代表性的Daphnilactone B类的虎皮楠生物碱
Figure 1 Some representative daphnilactone B-type alkaloids
表 1 Conia-ene反应条件优化
Table 1. Optimization of conia-ene reaction
| 序号 | 催化剂 | 添加剂 | 溶剂 | 产率/% |
| 1 | Au(PPh3)Cl (2 mol%) | AgBF4 (2 mol%) | V(PhMe):V(MeOH)=10:1 | 44 |
| 2 | Au(PPh3)Cl (2 mol%) | AgBF4 (2 mol%) | V(DCM):V(H2O)=10:1 | 52 |
| 3 | Au(PPh3)NTf2 (2 mol%) | — | V(PhMe):V(MeOH)=10:1 | 62 |
| 4 | Au(PPh3)NTf2 (10 mol%) | — | V(PhMe):V(MeOH)=10:1 | 69 |
| 5 | Au(PPh3)NTf2 (10 mol%) | — | V(DCM):V(H2O)=10:1 | 95 |
| 6 | Au(PPh3)NTf2 (10 mol%) | — | V(丙酮):V(H2O)=10:1 | 57 |
| 7 | Au(PPh3)NTf2 (10 mol%) | — | V(丙酮):V(丙酸)=20:1 | 62 |
| 8 | Au(PPh3)NTf2 (10 mol%) | — | V(DCM):V(丙酸)=20:1 | 81 |
| 9 | Au(PPh3)NTf2 (10 mol%) | — | V(DCM):V(MeOH)=10:1 | 66 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们