
Scheme 1.
General methods for the synthesis of sec-alkylsilanes.
Mn-mediated reductive C(sp3)–Si coupling of activated secondary alkyl bromides with chlorosilanes
Liangliang Qi , Xiaobo Pang , Kai Yin , Qiu-Quan Pan , Xiao-Xue Wei , Xing-Zhong Shu

Alkylsilanes are key structural motifs in many functional molecules, and they have found broad applications in organic synthesis, material science, and medicinal chemistry [1-4]. The streamlined synthetic approaches to these compounds involve the treatment of alkyl Grignard/lithium reagents with chlorosilanes [5] and catalytic hydrosilylation of alkenes [6-10]. Despite formidable advances, several challenges remain, involving the construction of secondary alkylsilanes [11-16]. Transition-metal-catalyzed C–Si coupling offers an attractive solution to this problem [17-25]. For example, the groups of Oestreich and Fu and others have recently reported several elegant works on the synthesis of alkylsilanes by coupling of secondary alkyl electrophiles with silylmetallic species (e.g., Si–M, M = Li, B, Mg and Zn) (Scheme 1a, path a) [26-33]. Meanwhile, Watson and coworkers have achieved the couplings of alkyl Grignard and Zinc reagents with silyl electrophiles (Scheme 1a, path b) [34, 35]. Alternatively, the reaction of carbon and silyl electrophiles may deliver the same compounds by using more readily available chemicals, but it remains largely unexplored (Scheme 1a, path c).
The cross-electrophile reaction has recently emerged as a promising tool for the construction of C-C bonds [36-44]. This method has shown high step-economy, good functionality tolerance, and an orthogonal chemoselectivity to classic cross-couplings. Our group has an ongoing interest in the exploration of new coupling partners for this chemistry [45-50]. Very recently, we reported the first reductive C–Si coupling reaction, which allows for the synthesis of aryl and vinylsilanes from carbon electrophiles and chlorosilanes [51]. This method was later expanded to forming C(sp3)-Si bonds by Oestreich, Zhang, and our groups using nickel catalysis [52-54]. However, due to the low reactivity of Si–Cl towards transition metals [55-57], the nickel-catalyzed route appears to be highly dependent on vinyl chlorosilanes. Moreover, the reaction of secondary alkyl electrophiles remains largely unexplored, and current studies focus on the synthesis of α-cyano alkylsilanes (Scheme 1b) [52].
Herein, we report an Mn-mediated approach for reductive C(sp3)–Si bond formation. This protocol avoids the requirement for activation of Si–Cl by transition metals, and it thus allows for the synthesis of organosilanes using a wide range of chlorosilanes. The feasibility of this approach is illustrated by the coupling reaction of α-functionalized alkyl bromides with chlorosilanes (Scheme 1c).
α-Functionalized alkylsilanes are useful building blocks in organic synthesis [58-62]. The importance of these structural motifs has triggered continuing efforts for their synthesis. The general approaches to these compounds involve the nucleophilic substitution reactions using strong basic conditions (e.g., base = n-BuLi, LDA and NaHMDS) [63, 64] and carbenes/silenes insertion reactions [65-68]. The coupling method was realized by Oestreich et al., which allows for the synthesis of α-silyl nitriles/esters from α-triflyloxy nitriles/esters and silyl organometallic species [29] and chlorosilanes [52]. Considering the importance of phosphorus or sulfur in ligand design [69-71] and the broad application of silyl moieties in developing heterogeneous catalysts [72], we here report a new method for the synthesis of α-silyl organophosphorus and sulfones by reductive C(sp3)–Si bond-forming reaction.
We started our investigation by studying the reaction of α-bromo phosphorus 1a with chlorosilane 2a (Table 1). We found the treatment of them with Mn (3.0 equiv.) in 1, 4-dioxane/DMA (4:1, 0.1 mol/L) gave the desired product 3a in 65% isolated yield (entry 1). The nitrogen solvent is essential to the success of this reaction, but the addition of 1, 4-dioxane improved the yield from 53% to 69% (entries 1–3). Lowing the loading of Mn resulted in decreased yields of 3a; the use of Mn (4.0 equiv.) did not improve the reaction efficiency (entries 4–6). The use of other metal reductants also delivered the target product but gave relatively lower yields (entries 7–9). No reaction was observed in the presence of an organic reductant, TDAE, or in the absence of Mn (entries 10 and 11).
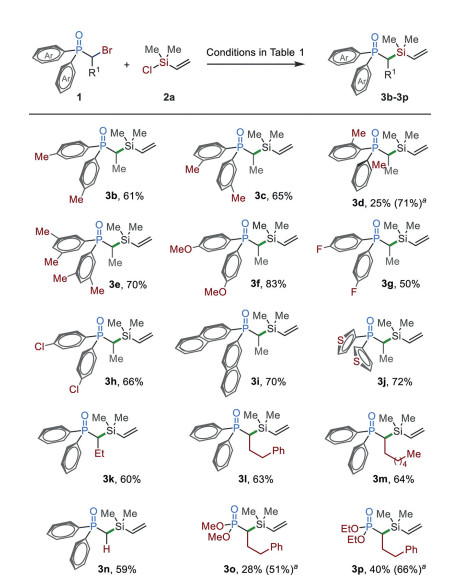
With the optimized reaction conditions in hand, we then studied the reactions of various α-bromo organophosphorus 1 with chlorosilane 2a (Scheme 2). Vinyl chlorosilane 2a was used as a coupling partner since this moiety has found promising applications in organic synthesis and material science [73-75]. Diarylphosphine oxides, either bearing an electron-rich (3b–3f) or electron-poor (3g and 3h) substituent, were coupled with 2a efficiently. Substituents at the para- and meta-positions were tolerated (3b and 3c). However, when an ortho-substituted substrate was used, the reaction gave a low yield of 3d. This challenge has been addressed by the addition of NiI2 (5 mol%), which significantly improved the yield from 25% to 71%. The reactions of naphthalenyl and heteroaryl substrates afforded 3i and 3j in good yields. The alkyl substituents could be expanded from methyl (3b–3j) and ethyl (3k) groups to long alkyl chains (3l and 3m). The reaction of primary alkyl bromide afforded silylated product 3n in 59% yield. In addition to phosphine oxides, the reactions of phosphonates also afforded the target products 3o and 3p in good yields in the presence of additional nickel.
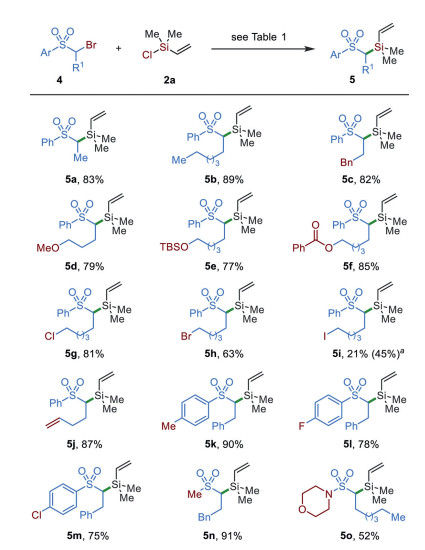
Slightly adjustment of reaction conditions, including the concentration (0.2 mol/L) and temperature (at 15 ℃), enables the silylation of α-bromo sulfones to be established (Scheme 3). The presence of different alkyl groups was tolerated (5a–5j). Functionalities, including ether (5d), silyl ether (5e), ester (5f), alkyl halides (5g–5i), and terminal alkene (5j), were tolerated. The scope of aryl sulfones was extended from electron-neutral (5a–5j) to electron-rich (5k) and –poor (5l and 5m) derivatives without the loss of reaction efficiency. The reactions of dialkyl sulfone and sulfamide also worked well and afforded silylation products 5n and 5o in 91% and 52% yields, respectively.
The substrate scope of the reaction with respect to chlorosilanes is shown in Scheme 4. The reactions of non-vinyl substituted chlorosilanes generally gave useful yields of target products. The use of 5 mol% of NiI2 significantly improved the reaction efficiency. The presence of a long alkyl chain and sterically hindered substituent were tolerated (3q and 3r). Aryl chlorosilanes, including PhMe2SiCl, Ph2MeSiCl, and (C6F5)Me2SiCl, also worked well; the reactions afforded target products 3s–3u in good yields. Functionalized dimethyl chlorosilanes, such as those bearing nitrile (3v), allyl (3x), and chloride (3y) groups, were also tolerated.

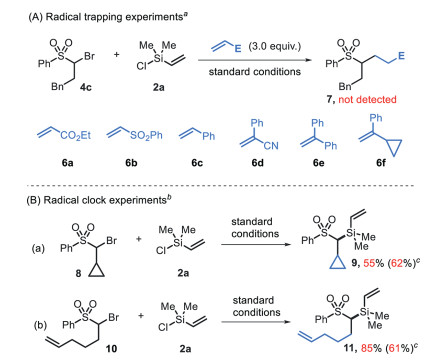
To insight into the mechanism of this process, several control experiments were performed. (1) In the presence of Michael acceptors 6a-6f (3.0 equiv.), the reactions proceeded well to afford the desired product in 58%−76% yields (see Scheme S1 in Supporting information for details), and they did not result in any radical trapping product 7 (Scheme 5A). (2) The reaction of α-cyclopropyl substrate 8 afforded the directly coupling product 9 in 55% yield with no ring-opening derivative was observed (Scheme 5B-a). (3) The reaction of bromo(pent-4-en-1-yl)sulfone gave silylated product 11 in 85% yield and did not result in any cyclized side-product (Scheme 5B-b). Similar results were also obtained in the presence of nickel (Scheme 5B). These results suggest that a free radical is not generated in this process.
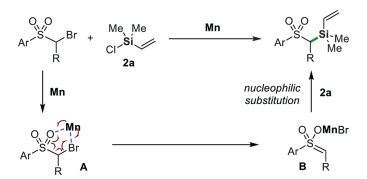
Based on the above results and literature reports, we tentatively proposed a mechanism for Mn-mediated C(sp3)–Si bond-forming reaction (Scheme 6). The coordination of α-bromo substrate to Mn gives complex A, which may undergo a concerted oxidative addition reaction to afford complex B. The nucleophilic substitution of this complex with chlorosilanes affords the desired product [63, 64, 76-78]. The formation of enol intermediate B is consistent with the deuterium experiments; in the presence of D2O (1.0 equiv.), the reaction of alkyl-Br 4c and 2a gave alkyl-D in 75% yield and a trace of alkyl-Si 5c (Scheme S2 in Supporting information). At present, the effect of nickel is unclear. We suppose it might be reduced to low-valent species like nanoparticle, which promote the reaction similar to the process of Mn.
In conclusion, we have reported a reductive C(sp3)–Si bond-forming reaction of secondary alkyl bromides with chlorosilanes and thus established a new method for the synthesis of α-silylated organophosphorus and sulfones. This ligand-free reaction proceeds under mild conditions and tolerates various functional groups. Further expansion of the scope of reductive C–Si coupling reactions are ongoing in our laboratory.
All authors declare no competing financial interests
We thank the National Natural Science Foundation of China for its financial support (No. 22071084).
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.03.070.
T. Hiyama, Organosilicon compounds in cross-coupling Reactions tamejiro hiyama, in: F. Diederich, P.J. Stang (Eds.), Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, New York, 1998, pp. 421–454.
T. Hiyama, M. Oestreich, Organosilicon Chemistry: Novel Approaches and Reactions, Wiley-VCH, Weinheim, 2019.
B. Boutevin, F. Guida-Pietrasanta, A. Ratsimihety, Silicon containing polymers, in: R.G. Jones, W. Ando, J. Chojnowski (Eds.), The Science and Technology of Their Synthesis and Application, Springer, Dordrecht, 2000, pp. 79–112.
R. Tacke, S. Dorrich, Drug design based on the carbon/silicon switch strategy, in: J. Schwarz (Ed.), Atypical Elements in Drug Design, Springer, Cham, 2016, pp. 29–59.
B.A. Keay, I. Fleming, Arylsilanes Science of Synthesis: Houben-Weyl Methods of Molecular Transformations, 4, Georg Thieme Verlag, 2002, pp. 685–712.
B. Marciniec, Hydrosilylation: A Comprehensive Review on Recent Advances, Springer, Dordrecht, 2008.
D. Troegel, J. Stohrer, Coord. Chem. Rev. 255 (2011) 1440–1459. doi: 10.1016/j.ccr.2010.12.025
X. Du, Z. Huang, ACS Catal. 7 (2017) 1227–1243. doi: 10.1021/acscatal.6b02990
J. Chen, J. Guo, Z. Lu, Chin. J. Chem. 36 (2018) 1075–1109. doi: 10.1002/cjoc.201800314
K. Li, M. Nie, W. Tang, Green Synth. Catal. 1 (2020) 171–174. doi: 10.1016/j.gresc.2020.08.003
S. Mallick, E.U. Wurthwein, A. Studer, Org. Lett. 22 (2020) 6568–6572. doi: 10.1021/acs.orglett.0c02337
X. Du, Y. Zhang, D. Peng, Z. Huang, Angew. Chem. Int. Ed. 55 (2016) 6671–6675. doi: 10.1002/anie.201601197
M.W. Gribble, M.T. Pirnot, J.S. Bandar, R.Y. Liu, S.L. Buchwald, J. Am. Chem. Soc. 139 (2017) 2192–2195. doi: 10.1021/jacs.6b13029
C. Wang, W.J. Teo, S. Ge, ACS Catal. 7 (2017) 855–863. doi: 10.1021/acscatal.6b02518
B. Cheng, P. Lu, H. Zhang, X. Cheng, Z. Lu, J. Am. Chem. Soc. 139 (2017) 9439–9442. doi: 10.1021/jacs.7b04137
M.Y. Hu, Q. He, S.J. Fan, et al., Nat. Commun. 9 (2018) 221–231. doi: 10.1038/s41467-017-02472-6
S. Bahr, W. Xue, M. Oestreich, ACS Catal. 9 (2019) 16–24. doi: 10.1021/acscatal.8b04230
K. Murakami, K. Hirano, H. Yorimitsu, K. Oshima, Angew. Chem. Int. Ed. 47 (2008) 5833–5835. doi: 10.1002/anie.200801949
M. Tobisu, Y. Kita, N. Chatani, J. Am. Chem. Soc. 128 (2006) 8152–8153. doi: 10.1021/ja062745w
V. Murugesan, V. Balakrishnan, R. Rasappan, J. Catal. 377 (2019) 293–298. doi: 10.1016/j.jcat.2019.07.026
Y.Y. Kong, Z.X. Wang, Adv. Synth. Catal. 361 (2019) 5440–5448. doi: 10.1002/adsc.201900949
K.M. Korch, D.A. Watson, Chem. Rev. 119 (2019) 8192–8228. doi: 10.1021/acs.chemrev.8b00628
J. Terao, K. Torii, K. Saito, et al., Angew. Chem. Int. Ed. 37 (1998) 2653–2656. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2653::AID-ANIE2653>3.0.CO;2-3
J.R. McAtee, S.E.S. Martin, D.T. Ahneman, K.A. Johnson, D.A. Watson, Angew. Chem. Int. Ed. 51 (2012) 3663–3667. doi: 10.1002/anie.201200060
S.E.S. Martin, D.A. Watson, J. Am. Chem. Soc. 135 (2013) 13330–13333. doi: 10.1021/ja407748z
C.K. Chu, Y. Liang, G.C. Fu, J. Am. Chem. Soc. 138 (2016) 6404–6407. doi: 10.1021/jacs.6b03465
W. Xue, Z.W. Qu, S. Grimme, M. Oestreich, J. Am. Chem. Soc. 138 (2016) 14222–14225. doi: 10.1021/jacs.6b09596
W. Xue, M. Oestreich, Angew. Chem. Int. Ed. 56 (2017) 11649–11652. doi: 10.1002/anie.201706611
J. Scharfbier, H. Hazrati, E. Irran, M. Oestreich, Org. Lett. 19 (2017) 6562–6565. doi: 10.1021/acs.orglett.7b03279
W. Xue, R. Shishido, M. Oestreich, Angew. Chem. Int. Ed. 57 (2018) 12141–12145. doi: 10.1002/anie.201807640
J. Scharfbier, B.M. Gross, M. Oestreich, Angew. Chem. Int. Ed. 59 (2020) 1577–1580. doi: 10.1002/anie.201912490
S. Wang, M. Sun, H. Zhang, et al., CCS Chem. 3 (2021) 2164–2173. doi: 10.31635/ccschem.020.202000447
V. Balakrishnan, V. Murugesan, B. Chindan, R. Rasappan, Org. Lett. 23 (2021) 1333–1338. doi: 10.1021/acs.orglett.0c04316
A.P. Cinderella, B. Vulovic, D.A. Watson, J. Am. Chem. Soc. 139 (2017) 7741–7744. doi: 10.1021/jacs.7b04364
B. Vulovic, A.P. Cinderella, D.A. Watson, ACS Catal. 7 (2017) 8113–8117. doi: 10.1021/acscatal.7b03465
C.E.I. Knappke, S. Grupe, D. Gärtner, et al., Chem. Eur. J. 20 (2014) 6828–6942. doi: 10.1002/chem.201402302
T. Moragas, A. Correa, R. Martin, Chem. Eur. J. 20 (2014) 8242–8258. doi: 10.1002/chem.201402509
E.L. Lucas, E.R. Jarvo, Nat. Rev. Chem. 1 (2017) 0065–0071. doi: 10.1038/s41570-017-0065
M.J. Goldfogel, L. Huang, D.J. Weix, Cross electrophile coupling: principles and new reactions, in: S. Ogoshi (Ed.), Nickel Catalysis in Synthesis: Methods and Reactions, Wiley-VCH, Weinheim, 2020, pp. 183–222.
J. Liu, Y. Ye, J.L. Sessler, H. Gong, ACC Chem. Res. 53 (2020) 1833–1845. doi: 10.1021/acs.accounts.0c00291
K.E. Poremba, S.E. Dibrell, S.E. Reisman, ACS Catal. 10 (2020) 8237–8246. doi: 10.1021/acscatal.0c01842
X. Pang, X. Peng, X.Z. Shu, Synthesis (Mass) 52 (2020) 3751–3763. doi: 10.1055/s-0040-1707342
P. Zheng, P. Zhou, D. Wang, et al., Nat. Commun. 12 (2021) 1646. doi: 10.1038/s41467-021-21947-1
D. Wang, T. Xu, ACS Catal. 11 (2021) 12469–12475. doi: 10.1021/acscatal.1c03265
X.G. Jia, P. Guo, J. Duan, X.Z. Shu, Chem. Sci. 9 (2018) 640–645. doi: 10.1039/C7SC03140H
X.B. Yan, C.L. Li, W.J. Jin, P. Guo, X.Z. Shu, Chem. Sci. 9 (2018) 4529–4534. doi: 10.1039/C8SC00609A
R.D. He, C.L. Li, Q.Q. Pan, et al., J. Am. Chem. Soc. 141 (2019) 12481–12486. doi: 10.1021/jacs.9b05224
H. Xie, J. Guo, Y.Q. Wang, et al., J. Am. Chem. Soc. 142 (2020) 16787–16794. doi: 10.1021/jacs.0c07492
P. Guo, K. Wang, W.J. Jin, et al., J. Am. Chem. Soc. 143 (2021) 513–523. doi: 10.1021/jacs.0c12462
P.F. Su, K. Wang, X. Peng, et al., Angew. Chem. Int. Ed. 60 (2021) 26571–26576. doi: 10.1002/anie.202112876
J. Duan, K. Wang, G.L. Xu, et al., Angew. Chem. Int. Ed. 59 (2020) 23083–23088. doi: 10.1002/anie.202010737
L. Zhang, M. Oestreich, Angew. Chem. Int. Ed. 60 (2021) 18587–1859. doi: 10.1002/anie.202107492
M. Xing, H. Cui, C. Zhang, Org. Lett. 23 (2021) 7645–7649. doi: 10.1021/acs.orglett.1c02887
J. Duan, Y. Wang, L. Qi, et al., Org. Lett. 23 (2021) 7855–7859. doi: 10.1021/acs.orglett.1c02874
H. Yamashita, M. Tanaka, M. Goto, Organometallics 16 (1997) 4696–4707. doi: 10.1021/om970214y
B. Vulovic, A.P. Cinderella, D.A. Watson, ACS Catal. 7 (2017) 8113–8117. doi: 10.1021/acscatal.7b03465
K. Matsumoto, J. Huang, Y. Naganawa, et al., Org. Lett. 20 (2018) 2481–2484. doi: 10.1021/acs.orglett.8b00847
D.J. Ager, The peterson olefination reaction, in: L.P. Paquette (Ed.), Organic Reactions, Wiley, 1990, pp. 1–219.
P.S. Jones, S.V. Ley, N.S. Simpkins, A.J. Whittle, Tetrahedron 42 (1986) 6519–6534. doi: 10.1016/S0040-4020(01)88114-X
M.B. Anderson, P.L. Fuchs, J. Org. Chem. 55 (1990) 337–342. doi: 10.1021/jo00288a058
D.J. Ager, J. Chem. Soc. Chem. Commun. (1984) 486–488.
D. Craig, S.V. Ley, N.S. Simpkins, G.H. Whitham, M.J. Prior, J. Chem. Soc. Perkin Trans. 1 (1985) 1949–1952.
E.E. Aboujaoude, S. Liétjé, N. Collignon, M.P. Teulade, P.A. Savignac, Synthesis 11 (1986) 934–937.
A.G. Shipov, Y.I. Baukov, Zh. Obshch. Khim. 54 (1984) 1842.
H. Keipour, V. Carreras, T. Ollevier, Org. Biomol. Chem. 15 (2017) 5441–5456. doi: 10.1039/C7OB00807D
W. Ando, A. Sekiguchi, T. Hagiwara, et al., J. Am. Chem. Soc. 101 (1979) 6393–6398. doi: 10.1021/ja00515a038
S.B.J. Kan, R.D. Lewis, K. Chen, F.H. Arnold, Science 354 (2016) 1048–1051. doi: 10.1126/science.aah6219
D. Chen, D.X. Zhu, M.H. Xu, J. Am. Chem. Soc. 138 (2016) 1498–1501. doi: 10.1021/jacs.5b12960
W. Tang, X. Zhang, Chem. Rev. 103 (2003) 3029–3070. doi: 10.1021/cr020049i
P.W.N.M. van Leeuwen, P.C.J. Kamer, C. Claver, O. Pamies, M. Dièguez, Chem. Rev. 111 (2011) 2077–2118. doi: 10.1021/cr1002497
M. Mellah, A. Voituriez, E. Schulz, Chem. Rev. 107 (2007) 5133–5209. doi: 10.1021/cr068440h
C. Li, Catal. Rev. 46 (2004) 419–492. doi: 10.1081/CR-200036734
L. Barfacker, D.E. Tom, P. Eilbraeht, Tetrahedron Lett. 40 (1999) 4031–4034. doi: 10.1016/S0040-4039(99)00678-4
P.P. Matloka, K.B. Wagener, J. Mol. Catal. A 257 (2006) 89–98. doi: 10.1016/j.molcata.2006.06.006
J.W. Park, C.H. Jun, J. Am. Chem. Soc. 132 (2010) 7268–7269. doi: 10.1021/ja102741k
M.I. Antczak, J.L. Montchamp, J. Org. Chem. 74 (2009) 3758–3766. doi: 10.1021/jo900300c
K. Chang, B. Ku, D.Y. Oh, Syn. Commun. 19 (1989) 1891–1898. doi: 10.1080/00397918908052580
M. Linnert, C. Bruhn, C. Wagner, D. Steinborn, J. Organomet. Chem. 691 (2006) 2358–2367. doi: 10.1016/j.jorganchem.2005.12.048

Scheme 2 The substrate scope of α-bromo organophosphorus. The reaction conditions as shown in Table 1. Bromide 1 (0.2 mmol) and chlorosilanes 2a (0.34 mmol) were used; isolated yields are given. a NiI2 (5 mol%) was added.

Scheme 3 The substrate scope of α-bromo sulfones. The reaction conditions as shown in Table 1; bromide 4 (0.2 mmol) and chlorosilane 2a (0.28 mmol) were used; reaction at 15 ℃ for 20 h; isolated yields are given. aNiI2 (5 mol%) was added.

Scheme 4 The substrate scope of chlorosilanes. The reaction conditions as shown in Table 1; bromide 1a (0.2 mmol) and chlorosilanes 2 (0.34 mmol) were used; isolated yields are given. aNiI2 (5 mol%) was added.

Scheme 5 Mechanistic studies. aReaction conditions as shown in Scheme 3; bromide 4 (0.2 mmol), chlorosilane 2a (0.28 mmol), and Michael acceptor 6a-6f (0.6 mmol) were used. bBromides 8/10 (0.2 mmol) and 2a (0.34 mmol) were used, isolated yields were given. cNiI2 (5 mol%) was added.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
