
Scheme 1.
Background and project synopsis of organic anion photosensitizers.
Phenylhydrazone anions excitation for the photochemical carbonylation of aryl iodides with aldehydes
Lei Shen , Yang Zhang , Linlin Zhang , Chuanwang Liu , Zhixian Ma , Kangjiang Liang , Chengfeng Xia

The visible light-induced photochemistry provides effective and powerful strategy for activation of organic molecules under mild conditions [1-10]. This trend was boosted by the tremendous identification and use of a variety of ruthenium and iridium complexes as well as organic dyes as photosensitizers for organic photosynthesis [11-21]. The special spectroscopic behavior of photoexcited organic anions has been reported more than half century ago [22,23]. Upon deprotonation, the absorption spectra of an organic anion is usually significant bathochromic shift, that provides the opportunity to absorb visible light and act as particularly potent electron donors from their photoexcited states [24-26]. Previous investigations of organic anions spectroscopic mainly focused on the hydrocarbon anions, which were generated under strong basic conditions and were very unstable for the development of photoreactions. Although only limited organic anions-based visible light-induced photosensitizer have reported up to date, they have displayed as potent entrants in photochemistry [26-30]. The anionic 9-anthrolates were also established as an efficient photosensitizer in the visible light-induced C—H carboxylation of (hetero)arenes with CO2 (Scheme 1A) [31,32]. Our research group developed a phenolate derivative, 2, 6-diisobutyl-4-phenylphenol (DBPP), as powerful photosensitizer with estimated excited-state reducing potential as low as −3.16 V vs. SCE, allowing the catalyzed reduction of aryl and alkyl chlorides and mesolytic cleavage of C–N and C–O bonds for the debenzylation (Scheme 1A) [33-35].
Hydrazones are highly important organic molecules because of their versatile synthetic utility in organic chemistry [36-43]. They serve as key building blocks or reagents in many chemical transformations such as Bamford-Stevens olefination [44-47] or carbonyl umpolung [48]. Hydrazones are also used as sources for the generation of nitrogen-centered radicals via oxidative deprotonation electron transfer by combination with various photosensitizers [49,50]. The proton of hydrazine could be deprotonation under basic conditions, thus enabling the possible bathochromic shift to visible spectra. The N-tosylhydrazine-derived hydrazone anion was firstly demonstrated for the direct photoactivation under sunlight irradiation. However, due to the high reductive potential of N-Ts-hydrazone anion, only oxidation by atmospheric oxygen was reported and the generated radical was limited in the intermolecular cyclization (Scheme 1B, left) [51,52]. Although the benzaldehyde hydrazone anion could not bathochromic shifted to visible spectra, Li and coworkers discovered that adding iodobenzene into the mixture generated a new absorption peak at 460 nm in UV–vis absorption spectrometry by formation an electron donor–acceptor (EDA) complex (Scheme 1B, right) [53].
Herein, we discovered that when the hydrazone was formed by a phenylhydrazine with arylaldehyde, the generated phenylhydrazone anion significantly shifted to visible light spectra without the need of formation EDA complex. Moreover, the photoexcited phenylhydrazone anion displayed strong reductive potential. With advantages of these discoveries, the photoexcited phenylhydrazone anion was explored to serve as a photosensitizer for the carbonylation of aryl halides through a SET process (Scheme 1C). Previously, the coupling of aldehydes with aryl halides were mainly achieved by transitional-metal catalysts [54-58]. This protocol offers a mechanistically distinguished pathway under mild conditions.
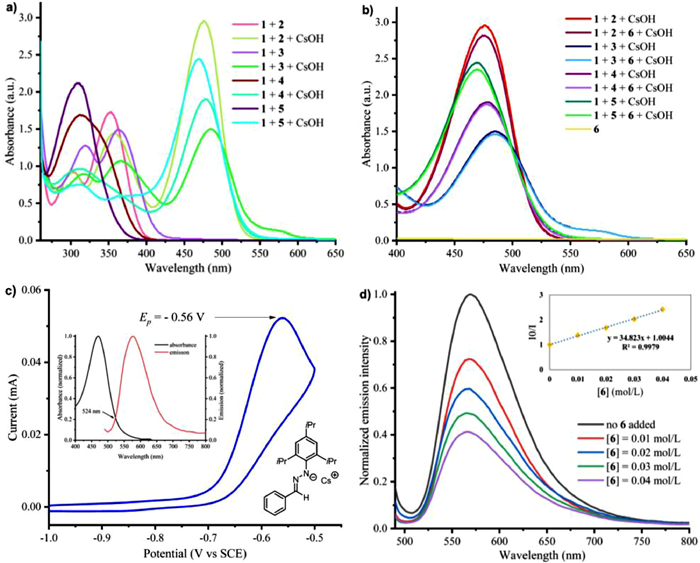
We observed that when phenylhydrazones, in-situ formed by mixing benzaldehyde 1 with different phenylhydrazines (2–5), were treated with CsOH·H2O, the corresponding spectra showed significant bathochromic shifts with absorption at 400−450 nm region (Fig. 1a, blue line). These phenomena indicated that the phenylhydrazine-derived hydrazone itself could absorb visible light. Moreover, we did not observe any changes in the ultraviolet-visible spectrum after p-iodotoluene 6 was added to the solution of the hydrazone anion (Fig. 1b, overlapped with the absorption of the hydrazine anion). These results were different from Li's spectra [53] about the formation of a ground-state EDA complex between the hydrazone anion with p-iodotoluene 6 [30,59-61]. Measurement of the fluorescence emission of deprotonated hydrazone under blue light irradiation showed that hydrazone anions were photoexcited. The redox potential of the excited hydrazone anion was estimated to be −2.93 V vs. SCE based on electrochemical and spectroscopic measurements (Fig. 1c, with hydrazine 5, other hydrazines see Supporting information) [62,63]. According to the reductive potentials of aryl iodides, we hypothesized that they were able to be reduced to aryl radicals by the photoexcited hydrazone via SET process. The hypothesis was supported from Stern–Volmer quenching studies (Fig. 1d), in which the excited state of the hydrazone anion was effectively quenched by p-iodotoluene 6.
To test the feasibility of our hypothesis, we started investigation with phenylhydrazones, in-situ generated by condensation of benzaldehyde 1 with different phenylhydrazines in DMSO, for the proposed carbonylation. After CsOH·H2O and p-iodotoluene 6 was added, the resulting mixture was irradiated with two 20 W blue light emitting diode (LED) lamps under argon for 24 h at room temperature. After the p-iodotoluene 6 was consumed as monitoring by TLC, the mixture was continued stirring under air atmosphere for additional 2 h. We found that when phenylhydrazine 2 was used for hydrazone formation, the carbonylation product 7 was isolated in 64% yield (Table 1, entry 1). Meanwhile, a p-tolyl-derived hydrazine by-product, formed by addition of p-tolyl radical to the benzene ring of phenylhydrazine, was identified. To avoid the undesired competitive radical pathway, phenylhydrazines 3–5 with substitutions on ortho or/and para-position, the p-tolyl radical addition sites, were then exploited (entries 2–4). The phenylhydrazine 5, which was substituted with tri-isopropyl groups on both ortho and para-positions, exhibited the most efficient carbonylation with 82% yield. Other bases, such as KOH, NaOH, K2CO3, DBU, and TMG, were also evaluated for the photochemical carbonylation (entries 5–9). Moderate yields were obtained when KOH and NaOH were used as base, while no reaction occurred with K2CO3 and DBU. The generation of hydrazone anion was essential for the photochemical carbonylation was further confirmed in the absence of any base (entry 10). Several solvents were also examined for the efficiency and the DMSO was discovered as the optimal solvent (entries 11−13). Decreasing the ratio of aldehyde 1 with iodotoluene 6 from 2:1 to 1:1 resulted in much lower yield, while increasing their ration to 3:1 slightly improved the yield (entries 14 and 15). The photochemical nature of the carbonylation was supported by the controlled experiment which was conducted in the dark (entry 16). A 2.0 mmol scale reaction was also investigated to prove the practicality of the arylation reaction, obtaining product 7 in 67% yield after 72 h.
With optimal conditions established, we then explored the scope of aryl iodides for photochemical carbonylation. As shown in Scheme 2, the aryl iodide with methyl group on ortho or para-position gave moderated yields too (8 and 9). Meanwhile, iodobenzenes substituted with methoxyl group on the ortho-, para-, and meso–positions were then examined under the photochemical conditions (10−12). Besides the iodotoluene, the bromotoluene was evaluated for the carbonylation, but resulting in much lower efficiency (12). Other ether and amine substituted substrates were also conducted to deliver the carbonylation products in moderate yields (13−19). It was discovered that only 45% yield was achieved for substrate with electron-withdrawing group (20). We reasoned that the electron-withdrawing group might lower the activity of decomposition from hydrazone to ketone under air conditions. The polycyclic aromatic iodides were then evaluated and found that they were suitable substrates for the photochemical carbonylation (21−23). Furthermore, the reaction showed good tolerance towards heterocycles, such as indole (24), benzofuran (25), benzothiophene (26), and thiophene (27).

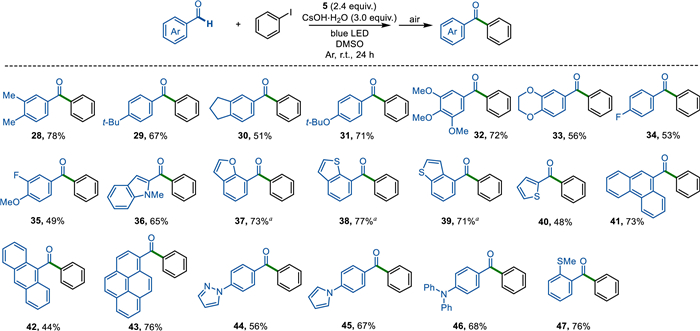
Subsequently, we extended the substrate scope to aryl aldehydes (Scheme 3). Under the standard conditions, the reaction of iodobenzene with different alkyl substituents on the aryl ring, including methyl (28), tert–butyl (29), and cyclopentyl moieties (30), proceeded smoothly and gave the carbonylation products in good yields. In addition, this photochemical protocol was also extended to various ether substituted aldehydes (31−33). Similarly, the electron-withdrawing group were also unfavorable for the conversion of hydrazones to ketones under air conditions (34 and 35). The hetero aromatic aldehydes and poly aromatic aldehydes were effectively employed as suitable substrates for this transformation and produced the corresponding products in moderate yields (36−43). Benzaldehydes with azo-heterocyclic (pyrazole and pyrrole) and hetero (amine and thioester) subsitutions were also discovered to be tolerated with moderate yields (44−47). However, this protocol was not practical to aliphatic aldehydes because the in-situ generated hydrazones did not bathochromically shifted enough to visible light region.
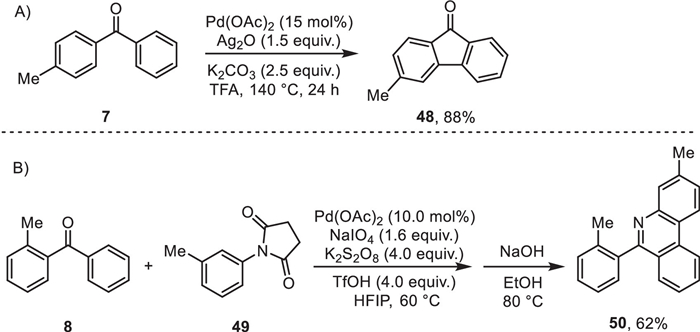
To expand the synthetic value of this method, we investigated the further chemical manipulation of synthesized carbonylation products. A dehydrogenative cyclization occurred by heating benzophenone 7 in the presence of Pd(OAc)2 and Ag2O to deliver fluorenone 48 in excellent yield (Scheme 4A) [64]. In addition, the ketone 8 was effectively transformed into the phenanthridine 50, an additional structural motif that can be easily found in many natural products and bio-active compounds, through a one-pot cascade cross-dehydrogenative-coupling/cyclization (Scheme 4B) [65].
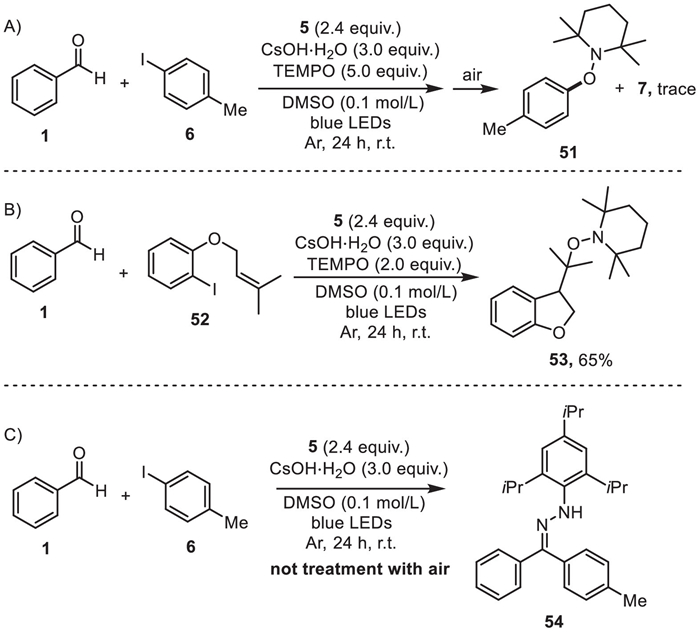
The electrochemical and spectroscopic experiments have suggested that the reduction of iodotoluene 6 (−2.35 V vs. SCE in DMSO, Fig. S8 in Supporting information) by the excited hydrazone anion (−2.93 V vs. SCE in DMSO) was thermodynamically feasible (Fig. 1c). Stern-Volmer quenching studies also revealed that the excited state of the hydrazone anion was effectively quenched by the iodotoluene 6 (Fig. 1d). To shed light on the mechanism of this photochemical transformation, several controlled experiments were then conducted. The addition of (2, 2, 6, 6-tetramethylpiperidin-1-yl)oxyl (TEMPO) strongly inhibited the photochemical arylation reaction, revealing the radical pathway of this protocol. Moreover, a TEMPO-trapped compound 51 was detected by HRMS, indicating the involvement of tolyl radical in the reaction (Scheme 5A) [66-69]. The generation of aryl radical was further confirmed when the iodobenzene 52 was armed with an isopentyl ether moiety. The exocyclic product 53 was obtained through a 5-exo-trig radical cyclization (Scheme 5B) [70]. The radical coupling intermediate 54 was detected by HRMS before the reaction mixture exposure to air for conversion to ketone (Scheme 5C) [71-73].
Based on the above observations, a plausible mechanism is outlined in Scheme 6. The in-situ generated hydrazone A was deprotonated by CsOH·H2O to produce the hydrazone anion A. Under the irradiation, a SET process occurred between the electronically excited species A* (Ered (A•/A*) = −2.93 V vs. SCE) and the iodotoluene 6 (Ered = −2.35 V vs. SCE, Fig. S8 in Supporting information) to give the hydrazone radical B and p-tolyl radical anion C, which was further converted to radical D after fragment. The intermediate F, a tautomer of compound 54, might be formed by direct coupling between the radical B and radical D. However, measurement of the quantum yield of the reaction was found to be Φ = 4.06 (Supporting information), suggesting a radical chain pathway involved. Therefore, it would be more reliable that p-tolyl radical D added with another molecule of hydrazone anion A to give the radical anion E, which is a strong single-electron reductant (the oxidation potential of E was estimated by measuring the reduction potential of 54, and a completely irreversible reduction wave was detected for 54 with Ered = −2.38 V vs. SCE in DMSO, Fig. S9 in Supporting information). This cyclic voltammetry results indicated that radical anion intermediate E could transfer an electron to iodotoluene 6 to regenerate p-tolyl radical D and propagate the radical chain [74].

In summary, this study established a redox-neutral photochemical carbonylation of aryl iodide by benzaldehyde at room temperature. Phenylhydrazones, which were in situ generated by condensation between benzaldehydes and phenylhydrazines, were firstly discovered to absorb visible light and act as new photosensitizer as well as reagent upon be deprotonated. Mechanistic studies revealed that the photo-activated phenylhydrazone was able to reduce aryl iodide to aryl radical via SET process. The aryl radical coupled with another molecule of phenylhydrazone anion to give a hydrazone radical anion intermediate. This intermediate was reductive enough to reduce another molecule of aryl iodide to aryl radical, resulting a radical chain process in this photochemical carbonylation. This protocol offers a convenient and efficient synthetic tool for accessing carbonylation products under redox neutral conditions, without the need of any transition metals.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 22271246 and 22101251) and Yunling Scholar Project of "Yunnan Revitalization Talent Support Program" for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
H. Wang, Y.M. Tian, B. König, Nat. Rev. Chem. 6 (2022) 745–755. doi: 10.1038/s41570-022-00421-6
K.P. Shing Cheung, S. Sarkar, V. Gevorgyan, Chem. Rev. 122 (2022) 1543–1625. doi: 10.1021/acs.chemrev.1c00403
S.L. Meng, C. Ye, X.B. Li, C.H. Tung, L.Z. Wu, J. Am. Chem. Soc. 144 (2022) 16219–16231. doi: 10.1021/jacs.2c02341
Y.Z. Cheng, Z. Feng, X. Zhang, S.L. You, Chem. Soc. Rev. 51 (2022) 2145–2170. doi: 10.1039/c9cs00311h
F.D. Lu, J. Chen, X. Jiang, et al., Chem. Soc. Rev. 50 (2021) 12808–12827. doi: 10.1039/d1cs00210d
J.D. Bell, J.A. Murphy, Chem. Soc. Rev. 50 (2021) 9540–9685. doi: 10.1039/d1cs00311a
G.E.M. Crisenza, D. Mazzarella, P. Melchiorre, J. Am. Chem. Soc. 142 (2020) 5461–5476. doi: 10.1021/jacs.0c01416
L. Marzo, S.K. Pagire, O. Reiser, B. König, Angew. Chem. Int. Ed. 57 (2018) 10034–10072. doi: 10.1002/anie.201709766
J.R. Chen, X.Q. Hu, L.Q. Lu, W.J. Xiao, Acc. Chem. Res. 49 (2016) 1911–1923. doi: 10.1021/acs.accounts.6b00254
D.M. Schultz, T.P. Yoon, Science 343 (2014) 985.
S.P. Pitre, L.E. Overman, Chem. Rev. 122 (2022) 1717–1751. doi: 10.1021/acs.chemrev.1c00247
N. Holmberg-Douglas, D.A. Nicewicz, Chem. Rev. 122 (2022) 1925–2016. doi: 10.1021/acs.chemrev.1c00311
A. Hossain, A. Bhattacharyya, O. Reiser, Science 364 (2019) eaav9713. doi: 10.1126/science.aav9713
K.L. Skubi, T.R. Blum, T.P. Yoon, Chem. Rev. 116 (2016) 10035–10074. doi: 10.1021/acs.chemrev.6b00018
M.H. Shaw, J. Twilton, D.W.C. MacMillan, J. Org. Chem. 81 (2016) 6898–6926. doi: 10.1021/acs.joc.6b01449
N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075–10166. doi: 10.1021/acs.chemrev.6b00057
C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322–5363. doi: 10.1021/cr300503r
H.Y. Song, Z.T. Zhang, H.Y. Tan, et al., Asian J. Org. Chem. 12 (2023) e202200658. doi: 10.1002/ajoc.202200658
D. Yang, Q. Yan, E. Zhu, J. Lv, W.M. He, Chin. Chem. Lett. 33 (2022) 1798–1816. doi: 10.1016/j.cclet.2021.09.068
N. Meng, Y. Lv, Q. Liu, R. Liu, X. Zhao, et al., Chin. Chem. Lett. 32 (2021) 258–262. doi: 10.1016/j.cclet.2020.11.034
Q.W. Gui, F. Teng, P. Yu, et al., Chin. J. Catal. 44 (2023) 111–116. doi: 10.1016/S1872-2067(22)64162-7
M.A. Fox, Chem. Rev. 79 (1979) 253–273. doi: 10.1021/cr60319a002
L.M. Tolbert, Acc. Chem. Res. 19 (1986) 268–273. doi: 10.1021/ar00129a002
E. Krogh, P. Wan, Top. Curr. Chem. 156 (1990) 93–116. doi: 10.1007/3-540-52379-0_4
J.P. Soumillion, Top. Curr. Chem. 168 (1993) 93–141. doi: 10.1007/3-540-56746-1_9
M. Schmalzbauer, M. Marcon, B. König, Angew. Chem. Int. Ed. 60 (2021) 6270–6292. doi: 10.1002/anie.202009288
E. Hasegawa, N. Izumiya, T. Miura, et al., J. Org. Chem. 83 (2018) 3921–3927. doi: 10.1021/acs.joc.8b00282
E. Hasegawa, Y. Nagakura, N. Izumiya, et al., J. Org. Chem. 83 (2018) 10813–10825. doi: 10.1021/acs.joc.8b01536
Q. Wang, M. Poznik, M. Li, P.J. Walsh, J.J. Chruma, Adv. Synth. Catal. 360 (2018) 2854–2868. doi: 10.1002/adsc.201800396
G. Filippini, M. Nappi, P. Melchiorre, Tetrahedron 71 (2015) 4535–4542. doi: 10.1016/j.tet.2015.02.034
M. Schmalzbauer, I. Ghosh, B. König, Faraday Discuss. 215 (2019) 364–378. doi: 10.1039/c8fd00176f
M. Schmalzbauer, T.D. Svejstrup, F. Fricke, et al., Chem 6 (2020) 2658–2672. doi: 10.1016/j.chempr.2020.08.022
K. Liang, Q. Liu, L. Shen, et al., Chem. Sci. 11 (2020) 6996–7002. doi: 10.1039/d0sc02160a
D. Wei, X. Li, L. Shen, et al., Org. Chem. Front. 8 (2021) 6364–6370. doi: 10.1039/d1qo01128f
K. Liang, X. Li, D. Wei, et al., Chem 9 (2023) 511–522. doi: 10.1016/j.chempr.2022.11.001
Z. Liu, P. Sivaguru, G. Zanoni, X. Bi, Acc. Chem. Res. 55 (2022) 1763–1781. doi: 10.1021/acs.accounts.2c00186
D. Zhu, L. Chen, H. Fan, Q. Yao, S. Zhu, Chem. Soc. Rev. 49 (2020) 908–950. doi: 10.1039/c9cs00542k
P. Xu, W. Li, J. Xie, C. Zhu, Acc. Chem. Res. 51 (2018) 484–495. doi: 10.1021/acs.accounts.7b00565
Y. Xia, J. Wang, Chem. Soc. Rev. 46 (2017) 2306–2362. doi: 10.1039/C6CS00737F
L.A. Tatum, X. Su, I. Aprahamian, Acc. Chem. Res. 47 (2014) 2141–2149. doi: 10.1021/ar500111f
A.R. Katritzky, L. Huang, M. Chahar, R. Sakhuja, C.D. Hall, Chem. Rev. 112 (2012) 1633–1649. doi: 10.1021/cr200076q
R. Lazny, A. Nodzewska, Chem. Rev. 110 (2010) 1386–1434. doi: 10.1021/cr900067y
M. Sugiura, S. Kobayashi, Angew. Chem. Int. Ed. 44 (2005) 5176–5186. doi: 10.1002/anie.200500691
W.R. Bamford, T.S. Stevens, J. Chem. Soc. (1952) 4735–4740. doi: 10.1039/JR9520004735(1952)4735–4740
P. Humphries, Bamford-Stevens reaction, in: J.J. Li (Ed.), Name Reactions for Homologations, John Wiley & Sons, Inc., 2009, pp. 642–652.
A.J. Clark, C.S. Guy, Reduction of Ketones to Alkenes, in: P. Knochel, G.A. Molander (Eds.), Comprehensive Organic Synthesis, 2nd Edition, Elsevier B.V., 2014, pp. 1143–1163.
M. Ghavre, Asian J. Org. Chem. 9 (2020) 1901–1923. doi: 10.1002/ajoc.202000336
S. Wang, B. König, Angew. Chem., Int. Ed. 60 (2021) 21624–21634. doi: 10.1002/anie.202105469
X.Y. Yu, Q.Q. Zhao, J. Chen, W.J. Xiao, J.R. Chen, Acc. Chem. Res. 53 (2020) 1066–1083. doi: 10.1021/acs.accounts.0c00090
M. Latrache, N. Hoffmann, Chem. Soc. Rev. 50 (2021) 7418–7435. doi: 10.1039/d1cs00196e
T. Zhang, Y. Meng, J. Lu, et al., Adv. Synth. Catal. 360 (2018) 3063–3068. doi: 10.1002/adsc.201701200
B. Sai Allaka, S. Basavoju, G.R. Krishna, Adv. Synth. Catal. 363 (2021) 3560–3565. doi: 10.1002/adsc.202100321
P. Pan, S. Liu, Y. Lan, H. Zeng, C.J. Li, Chem. Sci. 13 (2022) 7165–7171. doi: 10.1039/d2sc01909d
E.S. Isbrandt, A. Nasim, K. Zhao, S.G. Newman, J. Am. Chem. Soc. 143 (2021) 14646–14656. doi: 10.1021/jacs.1c05661
L. Wang, T. Wang, G.J. Cheng, et al., ACS Catal. 10 (2020) 7543–7551. doi: 10.1021/acscatal.0c02105
J.K. Vandavasi, X. Hua, H.B. Halima, S.G. Newman, Angew. Chem. Int. Ed. 56 (2017) 15441–15445. doi: 10.1002/anie.201710241
A. Bhattacharjya, P. Klumphu, B.H. Lipshutz, Nat. Commun. 6 (2015) 7401. doi: 10.1038/ncomms8401
S. Ko, B. Kang, S. Chang, Angew. Chem. Int. Ed. 44 (2005) 455–457. doi: 10.1002/anie.200462006
M. Silvi, E. Arceo, I.D. Jurberg, C. Cassani, P. Melchiorre, J. Am. Chem. Soc. 137 (2015) 6120–6123. doi: 10.1021/jacs.5b01662
A. Bahamonde, P. Melchiorre, J. Am. Chem. Soc. 138 (2016) 8019–8030. doi: 10.1021/jacs.6b04871
K. Liang, N. Li, Y. Zhang, T. Li, C. Xia, Chem. Sci. 10 (2019) 3049–3053. doi: 10.1039/c8sc05170d
M. Liang, Z.Y. Wang, L. Zhang, et al., Renew. Energy 36 (2011) 2711–2716. doi: 10.1016/j.renene.2011.03.003
L. Pause, M. Robert, J.M. Savéant, J. Am. Chem. Soc. 121 (1999) 7158–7159. doi: 10.1021/ja991365q
H. Li, R.Y. Zhu, W.J. Shi, K.H. He, Z.J. Shi, Org. Lett. 14 (2012) 4850–4853. doi: 10.1021/ol302181z
C. Zhang, T. Li, L. Wang, Y. Rao, Org. Chem. Front. 4 (2017) 386–391. doi: 10.1039/C6QO00522E
M. Kojima, K. Oisaki, M. Kanai, Chem. Commun. 51 (2015) 9718–9721. doi: 10.1039/C5CC02349A
W. Guo, L.Q. Lu, Y. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 2265–2269. doi: 10.1002/anie.201408837
Y. Iwata, Y. Tanaka, S. Kubosaki, T. Morita, Y. Yoshimi, Chem. Commun. 54 (2018) 1257–1260. doi: 10.1039/C7CC09140K
S.J. Kwon, H.I. Jung, D.Y. Kim, ChemistrySelect 3 (2018) 5824–5827. doi: 10.1002/slct.201801431
P. Bellotti, M. Koy, C. Gutheil, S. Heuvel, F. Glorius, Chem. Sci. 12 (2021) 1810–1817. doi: 10.1039/d0sc05551d
T. Tezuka, S. Ando, Chem. Lett. 14 (1985) 1621–1624. doi: 10.1246/cl.1985.1621
F. Sparatore, R. Cerri, J. Heterocycl. Chem. 16 (1979) 1001–1003. doi: 10.1002/jhet.5570160533
K. Livingstone, S. Bertrand, J. Mowat, C. Jamieson, Chem. Sci. 10 (2019) 10412–10416. doi: 10.1039/c9sc03032h
M.A. Cismesia, T.P. Yoon, Chem. Sci. 6 (2015) 5426–5434. doi: 10.1039/C5SC02185E

Figure 1 (a) UV–vis absorption spectra of DMSO solutions (10−4 mol/L) of aldehyde 1 with different hydrazines (2, 3, 4, and 5) with or without CsOH·H2O. (b) UV–vis absorption spectra of DMSO solutions (10−4 mol/L) of aldehyde 1, different hydrazines (2, 3, 4, and 5), CsOH·H2O, with or without aryl iodide 6. (c) Electrochemical and spectroscopic measurements of hydrazone (in-situ formed from aldehyde 1 and hydrazine 5). (d) Stern−Volmer fluorescence quenching experiment of hydrazone (in-situ formed from aldehyde 1 and hydrazine 5) with p-iodotoluene 6. PhCHO (1), PhNHNH2 (2), 4-MeOPhNHNH2 (3), 2, 4, 6-Me3-PhNHNH2 (4), 2, 4, 6-iPr3-PhNHNH2 (5), 4-MePhI (6).

Scheme 3 Substrate scope of aromatic aldehydes. a hydrazine 4 was used for the formation of hydrazones.

Scheme 6 Proposed mechanistic pathway for phenylhydrazone anion as photosensitizer and reagent.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:



 下载:
下载: