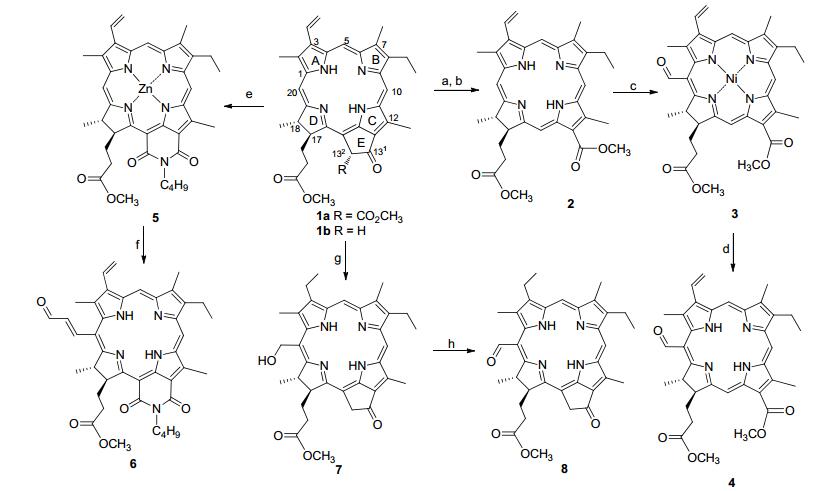
 图式1
单醛基取代叶绿素-a衍生物的合成
图式1.
Synthesis of mono-aldehyde group-substituted chlorophyll a derivatives
图式1
单醛基取代叶绿素-a衍生物的合成
图式1.
Synthesis of mono-aldehyde group-substituted chlorophyll a derivatives
引用本文:
姜齐永, 张珠, 刘洋, 姚楠楠, 王进军. 具有叶绿素碳架的二氢卟吩醛的合成及其与蛋白质的结合作用[J]. 有机化学,
2017, 37(7): 1814-1823.
doi:
10.6023/cjoc201610045

Citation: Jiang Qiyong, Zhang Zhu, Liu Yang, Yao Nannan, Wang Jinjun. Synthesis of Chlorin Aldehydes with Chlorophyllous Skeleton and Their Interactions with Protein[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1814-1823. doi: 10.6023/cjoc201610045
Citation: Jiang Qiyong, Zhang Zhu, Liu Yang, Yao Nannan, Wang Jinjun. Synthesis of Chlorin Aldehydes with Chlorophyllous Skeleton and Their Interactions with Protein[J]. Chinese Journal of Organic Chemistry, 2017, 37(7): 1814-1823. doi: 10.6023/cjoc201610045
具有叶绿素碳架的二氢卟吩醛的合成及其与蛋白质的结合作用
摘要:
以脱镁叶绿酸-a(b)甲酯为起始原料,经外接环修饰和色基金属化形成不同的叶绿素降解产物,再通过Vilsmeier酰基化和Blanc羟甲基化反应,在20-位上引进了新的官能基团.以四氧化锇、硝酸铊和空气为氧化剂,对二氢卟吩周环上的活性结构进行氧化,分别在二氢卟吩周环上的3-,7-和12-位以及外接环上引进了甲酰基和甲酰甲基,合成了一系列未见报道的叶绿素类二氢卟吩醛衍生物,其化学结构均经UV,IR,1H NMR及元素分析予以证实;同时也讨论了叶绿素类二氢卟吩醛基化的反应机理,并对新化合物与牛血清白蛋白的结合作用进行了研究.
English
Synthesis of Chlorin Aldehydes with Chlorophyllous Skeleton and Their Interactions with Protein
Abstract:
Pyropheophorbide-a (b) methyl esters were used as starting materials to form different chlorophyll degradation products by the modification of the exocyclic ring and the metallization of the chromophore. The new functional groups were introduced at 20-position via the Vilsmeier acylation and the Blanc hydroxymethylation. The active structures of chlorin peripheries were oxidized using osmium tetroxide, thaillum nitrate and air as oxidizing agent to introduce the formyl group and the formylmethyl group at 3-, 7-or 12-postion and on the exocyclic ring, respectively. A series of unreported chlorin aldehydes related to chlorophyll were synthesized and their chemical structures were characterized by elemental analysis, UV, IR and 1H NMR spectra. The reaction mechanisms on the hydroformylation for the chlorophyllous chlorins were discussed and the interactions of new compounds with bovine serum albumins were researched.
-
Key words:
- chlorophyll
- / chlorin
- / bovine serum albumins
- / chemical modification
- / florescence quenching
-
叶绿素-c和-d在3-和5-位上所连的甲酰基是区别于其他同系物的标识性结构, 其碳氧双键与二氢卟吩色基能够有效地形成广域的π电子体系, 对大环分子的光物理和光生物学性质可以产生特定的影响.除了用于天然叶绿素类族之间的转换之外, 甲酰基极易官能化的反应特征在合成新型四吡咯大环分子研究中也形成了独到的作用[1].因此, 在周环的不同位置上引进高反应性的醛基, 是设计具有应用价值的非对称性二氢卟吩的基本策略和重要的先期合成工作[2, 3].作为新的活性反应位点, 周环上碳氧双键的化学修饰和结构转换, 可以有效地构建和扩展各种碳架结构和改善分子的基本应用属性, 由此所得的新型叶绿素衍生物已经被广泛应用于光医学、超分子化学和分析化学等诸多方面[4~6].
叶绿素系列天然产物中的环上甲酰基所表现出的特殊作用启示我们进行新的尝试, 进一步深入对叶绿素化学的理论和应用研究.本工作以脱镁叶绿酸为起始原料, 利用其周环上的活性反应区域实施化学修饰, 通过多种常见的化学反应, 在四吡咯大环分子的不同位置上构建具有高反应活性的醛基, 为有效合成具有新颖结构和应用前景的叶绿素类二氢卟吩建立新的反应位点.同时, 通过荧光光谱法和分光光度法考察了部分新化合物与牛血清白蛋白(BSA)的结合作用及其在溶剂中的存在状态.
基于叶绿素周环上的芳香性含氮轮烯结构及其亲电反应的特点, 尝试利用Vilsmeier反应在20-位上选择性地引进甲酰基.最初曾直接对脱镁叶绿酸镍配合物进行甲酰基化, 由于周环上多处活性官能结构参与反应, 结果导致形成了难于分离的复杂混合物.因此, 我们从脱镁叶绿酸1a开始, 将原来外接环氧化、降解和重排反应合并[7], 一锅法生成二氢卟吩f-二酯(2, 32%).在三氯氧磷存在下, 其镍配合物在二氯甲烷中与N, N-二甲基甲酰胺顺利地发生Vilsmeier反应, 以48%的产率分离出甲酰基化二氢卟吩镍(3).后续的去金属化在浓硫酸中完成, 室温搅拌4 h后生成20-甲酰基二氢卟吩-f二酯(4, 44%).为了扩展Vilsmeier反应在叶绿素化学中的应用, 我们以红紫素-18二酰亚胺锌配合物5为电子给体, 选择3-二甲氨基丙烯醛和三氯氧磷为Vilsmeier试剂, 在反应过程中直接脱去中心金属离子, 以理想的产率得到20-甲酰乙烯基红紫素-18二亚酰胺(6).另一条C(20)-甲酰化的路线开始于焦脱镁叶绿酸(1b), 考虑到3-位乙烯基可能对后续反应形成不利影响, 先将其还原成乙基, 然后于多酸混合的溶液中进行羟甲基化反应, 以41%的产率得到20-羟甲基-meso-焦脱镁叶绿酸-a甲酯(7).再选用N-甲基吗啉氧化物(N-MMO)和高钌酸四丙基铵(TPAP)对其进行氧化, 分离出56%的20-甲酰基-meso-焦脱镁叶绿酸-a甲酯(8) (Scheme 1).
图式1
单醛基取代叶绿素-a衍生物的合成
图式1.
Synthesis of mono-aldehyde group-substituted chlorophyll a derivatives
建立多个活性反应区域是构建新颖化学结构的前提, 因而尝试在叶绿素二氢卟吩色基上实施双醛基化.以硝酸铊为氧化剂对7-位上已经连有甲酰基的脱镁叶绿酸-b甲酯的C(3)-乙烯基进行氧化, 高产率地生成3-二氧乙基脱镁叶绿酸-b甲酯(9).在88%的甲酸中对其缩醛结构进行水解, 生成78%的3-甲酰乙基脱镁叶绿酸-b甲酯(10); 另一双醛基化则从焦脱镁叶绿酸的空气氧化出发, 选择四氧化锇对所得的12-甲酰基二氢卟吩(11)的3-位乙烯基实施氧化和高碘酸钠裂解, 以中等产率分离出12-甲酰基焦脱镁叶绿酸-d甲酯(12); 利用焦脱镁叶绿酸外接环的氧化、成酐和酰胺化等一系列转化, 形成了N-羟乙基红紫素-18二酰亚胺(13), 经氢氧化锂促进下的空气氧化反应顺利地在12-位上形成甲酰基, 再通过混合氧化剂(N-MMO/TPAP)对12-甲酰基取代的红紫素-18二酰亚胺(14, 28%)的氮上的端位羟基进行氧化, 以50%的产率得到双醛基化的N-甲酰甲基-12-甲酰基红紫素-18二酰亚胺(15) (Scheme 2).
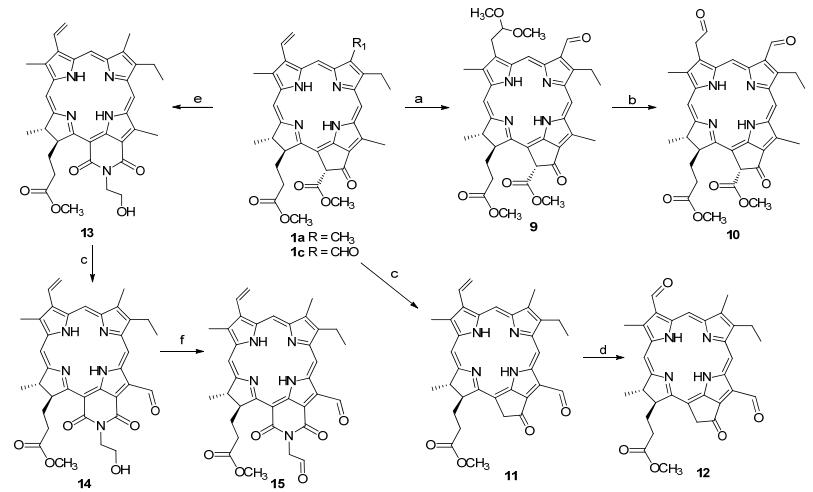 图式2
双醛基取代叶绿素-a衍生物的合成
图式2.
Synthesis of di-aldehyde group-substituted chlorophyll a derivatives
图式2
双醛基取代叶绿素-a衍生物的合成
图式2.
Synthesis of di-aldehyde group-substituted chlorophyll a derivatives
1 结果与讨论
1.1 叶绿素周环上的多电子性结构与醛基的形成
多电子性反应位点是形成碳氧双键的基本结构要求, 选择正确的亲电试剂和合适的反应条件则是顺利形成甲酰基的重要前提, 其多方面的选择性更是二氢卟吩官能化的重要的研究内容.叶绿素的基本碳架是由三个吡咯环和一个吡唑啉环桥连而成的二氢卟吩环系, 而大环中多种形式的芳香性氮杂轮烯结构基本决定了叶绿素衍生物的化学行为.例如, 焦脱镁叶绿酸-a甲酯(1b)的3-位乙烯基与苯乙烯相似, 其碳碳双键的π电子可以与多种氧化剂发生氧化反应, 因而也是在叶绿素衍生物碳架上形成醛基的最为便利的反应位点[8].
12-位甲基通过C-子环的β-位双键与外接E-环羰基可以形成插烯结构, 在酸或碱性条件下, 12-位甲基能够异构成多电子性的亚甲基而转化为烯醇式1c, 并与空气氧分子发生氧化而构建成新的碳氧双键.叶绿素二氢卟吩通过色基的异构存在着多个完整的吡咯子环, 除了大环分子的芳香性氮杂轮烯结构以外, 每个子环还要维持其自身的芳香性, 因而周环的电子密度将部分地离域到五元的吡咯环上, 导致meso-位的电子密度发生流失.由于5-和10-meso-位的两侧都连有吡咯环系结构, 所以, 处于吡咯和非芳香性含氮五元杂环的20-meso-位承载着相对大的电荷密度(图 1).
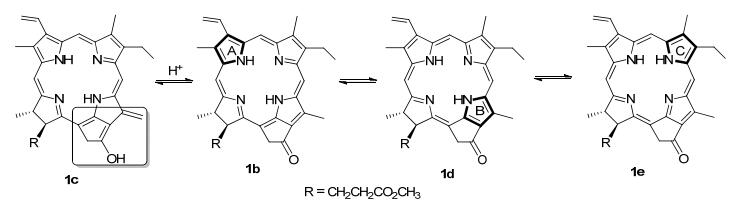 图 1
焦脱镁叶绿酸的色基结构互变
Figure 1.
Interconversion of the chromophore structure in pyropheophorbide
图 1
焦脱镁叶绿酸的色基结构互变
Figure 1.
Interconversion of the chromophore structure in pyropheophorbide
易于发生亲电取代是芳香性环系的基本反应特征, 根据前期对叶绿素类二氢卟吩的化学性质研究结果, 包括卤代、硝化、偶联、氰化和酰氧基化在内的取代反应, 均选择性地发生在20-meso-位上, 不同的亲电试剂所形成的反应结果也不尽相同, 亲电性较弱的试剂反应速度也相对较慢, 并且表现出可逆反应的特征[9].例如, 与重氮盐的偶联产物非常容易回归为起始原料, 而叶绿素降解产物的磺化反应由于逆向反应过快而一直未获成功[10].当meso-焦脱镁叶绿酸(6)与甲醛进行反应时, 首先与质子化甲醛的亲核加成生成大环鎓离子A, 20-meso-位质子离去后转化为羟甲基化二氢卟吩8', 再经混合氧化剂(N-MMO/TPAP)对其羟基实施氧化后得到20-甲酰基取代的二氢卟吩8.该反应在较强酸性和较高温度的条件下产率极低, 有时甚至分离不出期待产物; 如果将Blanc反应混合物放置24 h后再做分离, 同样也检测不到取代产物B.这些现象清晰地反映出该反应存在着明显的可逆反应特征(图 2).
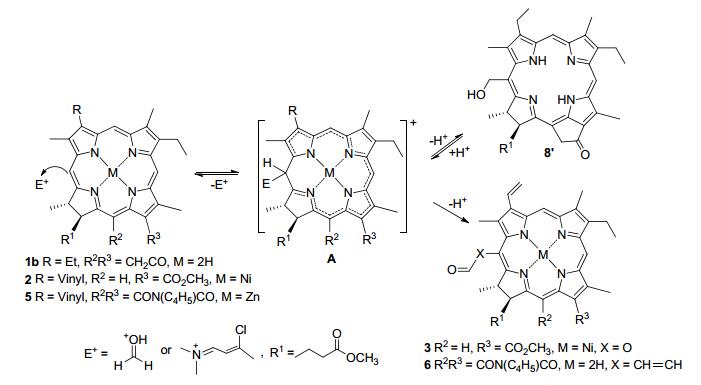 图 2
叶绿素二氢卟吩的20-位亲电取代反应
Figure 2.
The electrophilic substitution reactions of chlorophyllous chlorins at 20-position
图 2
叶绿素二氢卟吩的20-位亲电取代反应
Figure 2.
The electrophilic substitution reactions of chlorophyllous chlorins at 20-position
Vilsmeier试剂的亲电性能相对较低, 与二氢卟吩一般不发生亲电取代反应.如果将叶绿素降解衍生物转换成金属配合物, 镶入的中心镍或者锌离子不仅进一步促成大环分子的共平面, 其d轨道的非键电子也可以通过sp2杂化参与π体系的共轭, 从而有效地提高了二氢卟吩的芳香性和周环上的电子密度, 所生成的鎓离子A脱去质子后形成C(20)-甲酰化或者甲酰乙烯基化的产物3和6.与羟甲基化反应不同的是Vilsmeier产物在酸性条件下相当稳定, 其逆反应的趋势也非常小, 其20-位的酰基甚至在强酸剔除金属离子的过程中也保持不变.
1.2 叶绿素二氢卟吩醛与牛血清白蛋白的相互结合反应
与卟啉一样, 由于叶绿素类二氢卟吩大环上广域的π-π相互作用将导致其趋向聚集, 而聚集状态的改变能影响与生物体内靶分子的相互作用.在光学性质上, 卟啉聚集体可以表现出许多与单体状态不同的特征, 诸如荧光寿命的缩短、荧光的猝灭以及UV-Vis光谱中Soret带的减色等.因此, 可以通过测定叶绿素衍生物的光谱变化来考察其聚集状态, 并进一步分析和预测它们的光动力活性.
在蛋白质中, 由于色氨酸、酪氨酸的存在, 使其具有内源荧光(牛血清白蛋白λex/λem=255/340 nm), 而叶绿素衍生物在340 nm处没有荧光, 与BSA相互作用的聚合体也不显示荧光.以二氢卟吩醛11为例, 其与BSA的同步荧光谱图如图 3所示, 图 3清晰地显示, 随着叶绿素二氢卟吩浓度的增加, Δλ=15 nm与Δλ=60 nm的同步扫描荧光均呈现有规律的降低, 说明二氢卟吩对牛血清白蛋白的荧光有淬灭作用.
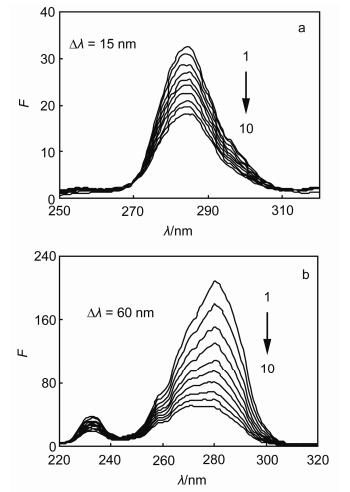 图 3
二氢卟吩11 (a)与BSA(b)的同步荧光图谱
Figure 3.
Synchronous fluorescence spectrum of 11 (a) and BSA (b)
图 3
二氢卟吩11 (a)与BSA(b)的同步荧光图谱
Figure 3.
Synchronous fluorescence spectrum of 11 (a) and BSA (b)
引起BSA荧光淬灭的原因有动态淬灭和(或)静态淬灭之分.动态猝灭由分子之间的碰撞引起, 而静态猝灭则是分子之间形成了没有荧光的配合物.以分子碰撞为主要荧光猝灭符合公式F0/F=1+k[Q][11], 其中, F0是淬灭剂不存在时的荧光强度, F为加入淬灭剂后的荧光强度, k为淬灭常数, [Q]为淬灭浓度.对于单一的动态淬灭或者静态淬灭过程, 以荧光分子的荧光强度变化F0/F[Q]对淬灭剂浓度作图应为直线关系, 其直线斜率则代表荧光淬灭过程速率常数k.由于生物大分子的最大动态淬灭过程的速率常数k<100 L•mol-1, 所以, 对于速率常数大于100 L•mol-1的荧光淬灭过程不可能是动态淬灭而只能是静态淬灭.
当小分子化合物与生物大分子之间存在结合作用时, 由参考文献[11]通过方程: lg[(F0-F)/F]=lg K0+nlg[Q]可计算叶绿素衍生物与BSA的结合常数, 其中F0是淬灭剂不存在时的荧光强度, F为加入淬灭剂后的荧光强度, K0为淬灭常数, [Q]为淬灭剂浓度, n为结合位点数.
表 1中的计算数据表明, 所选二氢卟吩与BSA形成了配合物, 存在较强的结合作用, 结合常数为5.40×104~3.94×106 L•mol-1, 结合位点数约等于1. 图 3中同步荧光扫描峰位和峰型均基本保持不变, 说明牛血清白蛋白中的酪氨酸与色氨酸残基所处的环境没有发生变化, 叶绿素二氢卟吩与BSA的作用没有影响到蛋白质的构象.尽管所选化合物的色基碳架不尽相同, 除了包括脱镁叶绿酸、二氢卟吩-f和红紫素-18类型的叶绿素降解产物以外, 还选择了相应的金属配合物, 但其与牛血清白蛋白的结合作用基本一致.
 表 1
lg(F0-F)/F与lg[Q]的方程、平衡常数K0和结合数n
Table 1.
Equation, K0 and n of lg(F0-F)/F vs. lg[Q]
表 1
lg(F0-F)/F与lg[Q]的方程、平衡常数K0和结合数n
Table 1.
Equation, K0 and n of lg(F0-F)/F vs. lg[Q]
叶绿素二氢卟吩 T/℃ lg(F0-F)/F vs. lg[Q] equation K0/(L·mol-1) n 1a 25 y=1.0457x+5.009 (R2=0.9954) 1.02×105 1.0457 37 y=1.1330x+5.6234 (R2=0.9974) 4.20×105 1.1330 3 35 y=1.2677x+5.9916 (R2=0.9966) 5.58×105 1.2677 37 y=0.9861x+4.8179 (R2=0.9959) 6.58×104 0.9861 11 25 y=1.2592x+6.5379 (R2=0.9977) 3.45×106 1.2592 37 y=1.2914x+6.5952 (R2=0.9902) 3.94×106 1.2914 13 25 y=1.1309x+5.5556 (R2=0.9954) 3.60×105 1.1309 37 y=0.9908x+4.7324 (R2=0.9902) 5.40×104 0.9908 2 结论
利用Vilsmeier和Blanc反应在叶绿素二氢卟吩的20-位上引进醛基和羟甲基结构, 再通过不同氧化剂的氧化反应, 将周环上原有和新建的活性结构转化成碳氧双键, 在叶绿素周环上的不同位置形成甲酰和甲酰甲基, 为进一步的结构转换提供了有效的活性反应位点, 拓展了在四吡咯大环上构建特殊碳架结构的切入手段, 为获取新型叶绿素类二氢卟吩提供了更加广泛的合成途径.所选二氢卟吩与牛血清白蛋白的结合作用表明, 不同色基结构的叶绿素二氢卟吩与BSA均可发生结合作用并导致其荧光静态淬灭, 与BSA的结合数也均等于1.
3 实验部分
3.1 仪器与试剂
元素分析用Perkin-Elmer 2400型元素分析仪测定; IR用Perkin-Elmer 1730型红外分光光度仪测定(KBr压片); UV-Vis用UV-160A型紫外分光光度计测定; 1H NMR用Brucker ARX-400型核磁共振仪测定, 内标为TMS.所用试剂均为分析纯或化学纯.脱镁叶绿酸-a甲酯(1a)、焦脱镁叶绿酸-a甲酯(1b)和脱镁叶绿酸-b甲酯(1c)按文献[12]制备.
3.2 二氢卟吩-f二酯(2) 的合成
将456 mg (0.752 mmol)脱镁叶绿酸甲酯-a 1溶解于50 mL二氯甲烷中, 加入由氢氧化钾饱和的5 mL甲醇溶液, 室温搅拌1 h, 减压除尽溶剂并将剩余物重新溶解于25 mL吡啶中, 加入15 mL 250 mg/mL的氢氧化钾甲醇溶液, 氮气保护, 搅拌回流3 h, 冷却至室温后加入30 mL水, 搅拌下滴加浓盐酸调节pH至3.加入30 mL二氯甲烷分出有机层, 水相用二氯甲烷萃取(40 mL×2), 合并有机层, 水洗两次, 用无水硫酸钠干燥, 减压除去溶剂, 用少量二氯甲烷将固体混合物溶解, 用重氮甲烷乙醚溶液甲基化, 再次除去溶剂, 将剩余物通过柱层析分离[洗脱剂: V(乙酸乙酯):V(正己烷)=1:4], 得136 mg绿色固体2 (0.241 mmol, 32%).其分析数据和物理常数与文献[7]一致.
3.3 20-甲酰基二氢卟吩-f二甲酯镍配合物(3) 的合成
将129 mg化合物2(0.228 mmol)溶解于30 mL吡啶中, 向反应体系加入0.8 g研细的醋酸镍, 于90 ℃搅拌反应6 h.减压除去反应溶剂, 将剩余物溶解于60 mL二氯甲烷中, 再加入60 mL水进行分层.分出有机相并水洗两次, 干燥后用重氮甲烷进行甲基化处理.减压除去溶剂, 将未分离的二氢卟吩-f二甲酯镍配合物溶解于20 mL二氯甲烷中待用. 0 ℃下向含有21 mg三氯氧磷(0.119 mmol)的反应瓶中滴加10 mg N, N-二甲基甲酰胺(0.114 mmol), 保持相同的温度搅拌反应15 min, 然后, 搅拌下将所得反应溶液加入已预冷却至0 ℃的含有镍配合物的二氯甲烷溶液中, 撤去冰水浴并室温搅拌18 h, 加入20 mL饱和的碳酸钠溶液, 再继续搅拌2 h.加入20 mL二氯甲烷分层, 用二氯甲烷萃取水相(20 mL×3), 合并有机相, 用无水硫酸钠干燥后用重氮甲烷甲基化, 减压除尽溶剂, 将剩余物通过柱层析分离[洗脱剂: V(乙酸乙酯):V(正己烷)=1:4]得71 mg亮绿色固体3(0.109 mmol, 48%). m.p. 183~186 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 408 (1.87×104), 507 (1.50×103), 538 (1.31×103), 609 (1.12×103), 668 (7.11×103) nm; 1H NMR (CDCl3) δ: 1.47 (t, J=7.6 Hz, 3H, 8-CH3), 2.29 (d, J=7.2 Hz, 3H, 18-CH3), 1.94~2.24 (m, 3H, 17a+17b-H), 2.30 (t, J=7.2 Hz, 1H, 17a+17b-H), 2.83, 2.91, 3.62, 3.65, 4.25 (each s, each 3H, CH3+OCH3), 3.19 (q, J=7.6 Hz, 2H, 8a-H), 4.00 (q, J=6.8 Hz, 1H, 18-H), 4.60 (dd, J=9.2, 1.6 Hz, 1H, 17-H), 5.94 (d, J=18.0 Hz, 1H, trans-3b-H), 5.96 (d, J=10.8 Hz, 1H, cis-3b-H), 7.43 (dd, J=18.0, 10.8 Hz, 1H, 3b-H), 7.75, 8.43, 9.86 (each s, each 1H, meso-H), 11.40 (s, 1H, 20-CHO); 13C NMR (CDCl3) δ: 198.2, 174.2, 172.9, 161.7, 157.8, 151.9, 149.6, 146.2, 143.1, 138.6, 137.9, 137.2, 137.1, 133.1, 129.7, 129.2, 128.6, 123.8, 114.3, 107.3, 105.5, 98.4, 93.9, 54.2, 51.6, 51.1, 34.4, 30.6, 30.3, 24.3, 21.8, 18.3, 15.0, 12.9, 12.1; IR (KBr) ν: 3446 (N—H), 2856 (C—H), 1740, 1710 (C=O), 1611 (C=C), 1531 (chlorin skeleton), 1463, 1307, 1263, 1132, 1089, 1041, 999 cm-1; EI-MS m/z: 652.3 (M+H+). Anal. calcd for C35H36N4NiO5: C 64.54, H 5.57, N 8.60; found C 64.39, H 5.62, N 8.49.
3.4 20-甲酰基二氢卟吩-f二甲酯(4) 的合成
将46 mg二氢卟吩镍配合物3 (0.071 mmol)溶解于6 mL浓硫酸中, 室温搅拌反应2 h, 用20%饱和的碳酸氢钠将所形成的反应溶液中和至pH约等于2, 加入100 mL二氯甲烷分层, 水洗有机层(100 mL×2), 用无水硫酸钠干燥后再经重氮甲烷甲基化, 浓缩后用硅胶柱层析分离[洗脱剂: V(石油醚):V(乙酸乙酯)=5:1], 得19 mg黑色固体4(0.031 mmol), 产率44%. m.p. 192~195 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 409 (1.58×104), 541 (9.28×102), 588 (8.27×102), 618 (7.11×102), 690 (1.23×104) nm; 1H NMR (CDCl3) δ: 0.04 (br s, 1H, NH), 0.13 (br s, 1H, NH), 1.70 (t, J=7.6 Hz, 3H, 8-CH3), 1.83 (d, J=7.0 Hz, 3H, 18-CH3), 1.95~2.08 (m, 1H, 17a+17b-H), 2.28~2.42 (m, 2H, 17a+17b-H), 2.53~2.72 (m, 2H, 17a+17b-H), 2.28, 3.21, 3.59, 3.61, 4.22 (each s, each 3H, CH3+OCH3), 3.69 (q, J=7.6 Hz, 2H, 8a-H), 4.43 (q, J=7.2 Hz, 1H, 18-H), 5.07 (d, J=10.1 Hz, 1H, 17-H), 5.18 (d, J=11.2 Hz, 1H, cis-3b-H), 6.32 (d, J=17.6 Hz, 1H, trans-3b-H), 7.95 (dd, J=17.6, 11.2 Hz, 1H, 3b-H), 8.56, 9.40, 9.62 (each s, each 1H, meso-H), 11.49 (s, 1H, 20-CHO); IR (KBr) ν: 3455 (N—H), 2958, 2867 (C—H), 1743, 1725 (C=O), 1642 (C=C), 1532 (chlorin skeleton), 1441, 1310, 1150, 104 cm-1; EI-MS m/z: 595.4 (MH+). Anal. calcd for C35H38N4O5: C 70.69, H 6.44, N 9.42; found C 70.59, H 6.37, N 9.31.
3.5 N-丁基红紫素-18二酰亚胺甲酯锌配合物(5) 的合成
将248 mg (0.409 mmol)脱镁叶绿酸甲酯-a (1a)溶解于25 mL二氯甲烷中, 加入由氢氧化钾饱和的5 mL甲醇溶液, 室温搅拌1.5 h, 加入20 mL水, 搅拌下滴加浓盐酸调节pH值约至2.加入30 mL二氯甲烷分出有机层, 水相用二氯甲烷萃取(40 mL×2), 合并有机层, 水洗两次后用无水硫酸钠干燥, 减压除尽溶剂, 用少量二氯甲烷将固体混合物溶解, 移至含有1.5 mL正丁胺和1 g研细的醋酸锌的20 mL干燥的吡啶中, 搅拌回流8 h.减压除去反应溶剂, 先后加入30 mL二氯甲烷和60 mL水进行分层.分出有机相并水洗两次, 干燥后用重氮甲烷进行甲基化, 减压除去溶剂, 将所得固体混合物经硅胶柱层析分离[洗脱剂: V(石油醚):V(乙酸乙酯)=3:1], 得194 mg绿色固体5 (0.278 mmol), 产率68%. m.p. 201~203 ℃; UV-vis (CH2Cl2)λmax [ε/(L• mol-1•cm-1)]: 418 (5.23×103), 483 (1.07×104), 513 (9.42×102), 551 (5.01×103), 709 (9.34×103) nm; 1H NMR (CDCl3) δ: 0.92 (t, J=6.6 Hz, 3H, N-C-C-C-CH3), 1.33 (t, J=7.6 Hz, 3H, 8a-CH3), 1.53 (d, J=7.2 Hz, 3H, 18-CH3), 1.24~1.64 (m, 4H, NC-CH2CH2-C), 1.88~2.01 (m, 2H, 17a+17b-H), 2.08~2.31 (m, 1H, 17a+17b-H), 2.52~2.73 (m, 1H, 17a+17b-H), 2.97, 3.00, 3.51, 3.61 (each s, each 3H, CH3+OCH3), 3.46 (q, J=7.6 Hz, 2H, 8a-H), 4.07 (q, J=7.2 Hz, 1H, 18-H), 4.27 (t, J=6.6 Hz, 2H, NCH2), 4.77 (dd, J=8.4, 3.0 Hz, 1H, 17-H), 5.95 (d, J=11.6 Hz, 1H, cis-3b-H), 5.97 (d, J=17.8 Hz, 1H, trans-3b-H), 7.60 (dd, J=17.8, 11.6 Hz, 3a-H), 7.93, 8.80, 9.04 (each s, 2H, meso-H); 13C NMR (CDCl3) δ: 175.1, 164.7, 156.0, 152.3, 150.7, 147.0, 144.0, 142.2, 141.3, 140.6, 140.2, 136.6, 136.2, 135.9, 134.7, 130.3, 129.9, 128.1, 124.9, 122.5, 113.9, 112.4, 109.9, 104.6, 101.3, 93.8, 52.7, 52.5, 49.8, 32.1, 30.7, 30.6, 24.5, 20.0, 18.2, 12.8, 12.3, 11.9; IR (KBr) v: 3410 (N—H), 2929 (C—H), 1726, 1690 (C=O), 1647 (C=C), 1551 (chlorin skeleton), 1437, 1380, 1271, 1141, 1070 cm-1; EI-MS m/z: 698.5 (M+H+). Anal. calcd for C38H41N5O4Zn: C 65.47, H 5.93, N 10.05; found C 65.51, H 6.09, N 10.19.
3.6 N-丁基20-(2’-甲酰乙烯基)红紫素-18二酰亚胺甲酯(6) 的合成
将0.4 mL三氯氧磷滴加到由0.4 mL 3, 3-二甲氨基丙烯醛和4 mL二氯甲烷组成的混合溶液, 在0 ℃下搅拌15 min.然后在相同温度下, 搅拌加入到已经预冷至-5 ℃的含有128 mg锌配合物5(0.184 mmol)的20 mL二氯甲烷中.撤去冰水浴, 室温搅拌18 h.搅拌下加入120 mL饱和碳酸氢钠水溶液, 室温搅拌过夜, 用二氯甲烷萃取反应液(30 mL×3), 合并有机层并水洗两次, 无水硫酸钠干燥后用重氮甲烷进行甲基化处理, 减压除去溶剂, 将所得固体混合物用硅胶柱层析分离[洗脱剂: V(石油醚):V(乙酸乙酯)=3:1], 得75 mg红色固体6 (0.108 mmol), 产率59%. m.p. 206~209 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 367 (5.93×103), 420 (1.12×104), 532 (9.41×102), 570 (5.08×103), 735 (9.45×103) nm; 1H NMR (CDCl3) δ: -0.02 (br s, 1H, NH), 0.60 (br s, 1H, NH), 0.88 (t, J=6.6 Hz, N-C-C-C-CH3), 1.30 (t, J=7.6 Hz, 3H, 8a-CH3), 1.63 (d, J=7.2 Hz, 3H, 18-CH3), 1.18~1.78 (m, 4H, NC-CH2CH2-C), 1.98~2.20 (m, 2H, 17a+17b-H), 2.27~2.50 (m, 1H, 17a+17b-H), 2.76~2.87 (m, 1H, 17a+17b-H), 2.98, 3.09, 3.62, 3.72 (each s, each 3H, CH3+OCH3), 3.56 (q, J=7.6 Hz, 2H, 8a-H), 4.34 (t, J=6.6 Hz, 2H, NCH2-), 4.61 (q, J=7.2 Hz, 1H, 18-H), 5.11 (dd, J=8.8, 2.7 Hz, 1H, 17-H), 6.12 (d, J=17.8 Hz, 1H, trans-3b-H), 6.16 (d, J=11.5 Hz, 1H, cis-3b-H), 6.19 (dd, J=15.7, 7.7 Hz, 1H, 20b-H), 7.71 (dd, J=17.8, 11.5 Hz, 1H, 3a-H), 8.93 (d, J=15.7 Hz, 1H, 20a-H), 9.30, 9.36 (each s, each 1H, meso-H), 10.11 (d, J=7.7 Hz, 20b-CHO); 13C NMR (CDCl3) δ: 201.1, 174.4, 172.8, 166.0, 161.7, 154.0, 151.3, 146.7, 144.7, 143.6, 140.7, 139.6, 137.1, 136.2, 135.7, 133.8, 131.0, 130.7, 130.2, 129.9, 123.2, 106.6, 106, 1, 101.8, 94.0, 52.6, 52.2, 49.8, 31.7, 31.2, 31.0, 30.7, 30.6, 26.1, 24.4, 24.3, 20.0, 18.2, 12.9, 12.8, .11.8; IR (KBr) v: 3346 (N—H), 2960 (C—H), 1738, 1704 (C=O), 1664 (C=C), 1542 (chlorin skeleton), 1458, 1365, 1174, 1068, 981 cm-1; EI-MS m/z: 688.2 (M+H+). Anal. calcd for C41H45N5O5: C 71.59, H 6.59, N 10.18; found C 71.61, H 6.44, N 9.99.
3.7 20-羟甲基-meso-焦脱镁叶绿酸-a甲酯(7) 的合成
将258 mg焦脱镁叶绿酸-a甲酯(1b, 0.470 mmol)溶解于20 mL二氯甲烷中, 搅拌下加入106 mg钯碳, 室温条件下通入氢气, 当反应体系的紫外可见光谱中在668 nm处的吸收峰消失后停止反应, 过滤, 用二氯甲烷洗涤, 合并滤液和洗涤液, 用无水硫酸钠干燥, 减压除尽溶剂.将所得固体混合物溶解于10 mL冰乙酸中, 搅拌下再加入6 mL浓磷酸和7 mL浓盐酸, 搅拌3 min后再慢慢滴加10 mL 40%的甲醛水溶液, 35 ℃下搅拌反应24 h.先后向反应体系加入40 mL水和40 mL二氯甲烷, 分出有机层, 水层用二氯甲烷萃取(30 mL×3), 合并有机层并水洗至中性, 用无水硫酸钠干燥, 除去95%的溶剂后用重氮甲烷甲基化, 减压浓缩, 剩余物经硅胶柱层析分离[洗脱剂: V(石油醚):V(乙酸乙酯)=2:1], 得到112 mg红色固体7 (0.193 mmol).产率41%. m.p. 213~215 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 412 (1.12×105), 510 (8.56×103), 544 (2.67×104), 609 (3.74× 104), 666 (6.34×104) nm; 1H NMR (CDCl3) δ: -1.34 (br s, 1H, NH), 0.70 (br s, 1H, NH), 1.50 (d, J=7.0 Hz, 3H, 18-CH3), 1.68 (t, J=7.6 Hz, 3H, 3a-CH3), 1.71 (t, J=7.6 Hz, 3H, 8a-CH3), 2.11~2.23 (m, 2H, 17a+17bH), 2.32~2.52 (m, 2H, 17a+17b-H), 3.66 (q, J=7.6 Hz, 2H, 8a-H), 3.84~3.92 (m, 2H, 3a-H), 3.27, 3.45, 3.54, 3.63 (each s, each 3H, CH3+OCH3), 4.20 (dd, J=8.2, 3.3 Hz, 1H, 17-H), 4.80 (q, J=7.2 Hz, 1H, 18-H), 5.13 (d, J=19.8 Hz, 1H, 132-H), 5.19 (d, J=19.8 Hz, 1H, 132-H), 6.01 (d, J=12.8, 1H, 20-CH2), 6.36 (d, J=12.8, 1H, 20-CH2), 9.32, 9.43 (each s, each 1H, meso-H); IR (KBr) v: 3469 (N—H), 2929 (C—H), 1747, 1699 (C=O), 1606 (C=C), 1534 (chlorin skeleton), 1457, 1311, 1171, 1143, 1020 cm-1; EI-MS m/z: 581.2 (M+H+). Anal. calcd for C35H40-N4O4: C 72.39, H 6.94, N 9.65; found C 72.19, H 7.06, N 9.77.
3.8 20-甲酰基-meso-焦脱镁叶绿酸-a甲酯(8) 的合成
在25 mL的干燥二氯甲烷中溶解139 mg二氢卟吩醇7 (0.239 mmol), 在氮气保护下, 迅速加入20 mg N-甲基吗啉氧化物(N-MMO)并避光搅拌15 min, 再小量分批加入30 mg高钌酸四丙基铵(TPAP), 搅拌6 h后反应结束.加入20 mL水, 静置过夜后分出水层, 水层用二氯甲烷萃取(15 mL×3), 合并有机层并用无水硫酸钠干燥.除去溶剂, 剩余物经硅胶柱层析分离[洗脱剂: V(石油醚):V(乙酸乙酯)=3:1], 得78 mg黄红色固体8 (0.134 mmol), 产率为56%. mp. 203~206 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 412 (1.38×105), 509 (2.02×104), 536 (2.81×104), 610 (3.06×104), 669 (5.78×104) nm; 1H NMR (CDCl3) δ: 0.04 (br s, 1H, NH), 0.88 (br s, 1H, NH), 1.42 (d, J=7.0 Hz, 3H, 18-CH3), 1.63 (t, J=7.6 Hz, 3H, 3a-CH3), 1.66 (t, J=7.6 Hz, 3H, 8a-CH3), 2.27~2.34 (m, 2H, 17a+17bH), 2.42~2.50 (m, 1H, 17a+17b-H), 2.57~2.65 (m, 1H, 17a+17b-H), 3.55 (q, J=7.6 Hz, 2H, 8a-H), 3.74 (q, J=7.5 Hz, 2H, 3a-H), 3.27, 3.45, 3.54, 3.63 (each s, each 3H, CH3+OCH3), 4.03 (dd, J=9.2, 3.2 Hz, 1H, 17-H), 4.98 (d, J=20.0 Hz, 1H, 132-H), 5.05 (d, J=20.0 Hz, 1H, 132-H), 5.18 (q, J=7.0 Hz, 1H, 18-H), 9.21, 9.27 (each s, each 1H, meso-H), 11.80(s, 1H, 20-CHO); IR (KBr) v: 3465 (N—H), 2920 (C—H), 1742, 1697 (C=O), 1604 (C=C), 1502 (chlorin skeleton), 1457, 1388, 1264, 1198, 1083, 1043 cm-1; EI-MS m/z: 579.4 (M+H+). Anal. calcd for C35H38N4O4: C 72.64, H 6.62, N 9.68; found C 72.81, H 6.76, N 9.60.
3.9 3-二氧乙基脱镁叶绿酸-b甲酯(9) 的合成
在50 mL二氯甲烷和25 mL甲醇混合液中溶解187 mg MPPb (1c, 0.301 mmol), 在0℃下将溶解于30 mL甲醇的150 mg三水硝酸铊(III)迅速加入到反应液中, 并在此温度下搅拌反应15 min, 然后加入30 mL亚硫酸氢钠甲醇饱和溶液, 室温搅拌20 min后滴加2 mL浓盐酸进行酸化, 将所产生的大量白色沉淀过滤, 分出有机层, 水层用二氯甲烷萃取(20 mL×3), 合并有机层并水洗一次, 用无水硫酸钠干燥后减压浓缩至干, 剩余物经硅胶柱层析[洗脱剂: V(石油醚):V(乙酸乙酯)=4:1], 得到148 mg黑绿色固体9 (0.217 mmol), 产率72%. mp. 197~199 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 418 (1.12×105), 484 (2.85×103), 512 (1.02×103), 552 (1.09×104), 666 (3.72×104) nm; 1H NMR (CDCl3) δ: -1.49 (br s, 1H, NH), 0.64 (br s, 1H, NH), 1.83 (t, J=7.6 Hz, 3H, 8-CH3), 1.81 (d, J=7.2 Hz, 3H, 18-CH3), 2.17~2.33 (m, 2H, 17a+17b-H), 2.46~2.88 (m, 2H, 17a+17b-H), 3.31, 3.53 (6H), 3.59, 3.67, 3.90 (each s, all 18H, CH3+OCH3), 4.05 (q, J=7.6 Hz, 2H, 8a-H), 4.11 (d, J=5.0 Hz, 2H, 3a-H), 4.18 (d, J=8.0 Hz, 1H, 17-H), 4.44 (q, J=7.0 Hz, 1H, 18-H), 5.09 (t, J=5.0 Hz, 1H, 3b-H), 6.22 (s, 1H, 132-H), 8.53, 9.56, 10.25 (each s, each 1H, meso-H), 11.15 (s, 1H, 7-CHO); IR (KBr) v: 3333 (N—H), 2851 (C—H), 1733, 1708 (C=O), 1614 (C=C), 1565 (chlorin skeleton), 1490, 1440, 1260, 1146, 1083, 1006 cm-1; EI-MS m/z: 683.4 (M+H+). Anal. calcd for C38H42N4O8: C 66.85, H 6.20, N 8.21; found C 66.71, H 6.47, N 8.35.
3.10 3-甲酰乙基脱镁叶绿酸-b甲酯(10) 的合成
将122 mg化合物9(0.179 mml)溶解于15 mL 88%的甲酸溶液中, 室温搅拌6 h, 加入20 mL水和35 mL二氯甲烷, 分出有机层, 水层用二氯甲烷萃取(15 mL×3), 合并有机层并水洗一次, 用无水硫酸钠干燥.减压浓缩, 剩余物硅胶柱层析分离[洗脱剂: V(石油醚):V(乙酸乙酯)=4:1], 得到89 mg暗红色固体10 (0.139 mmol), 产率78%. mp. 215~217 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 372 (1.58×103), 434 (9.87×104), 505 (2.14×103), 592 (3.95×103), 646 (4.24×103) nm; 1H NMR (CDCl3) δ: -1.47 (br s, 1H, NH), 0.64 (br s, 1H, NH), 1.84 (t, J=7.6 Hz, 3H, 8-CH3), 1.86 (d, J=7.2 Hz, 1H, 18-CH3), 2.23~2.41 (m, 2H, 17a+17b-H), 2.50~2.74 (m, 2H, 17a+17b-H), 4.06 (q, J=7.6 Hz, 2H, 8a-H), 3.32, 3.62, 3.91, 3.94 (each s, each 3H, OCH3+CH3), 4.24 (d, J=8.8 Hz, 1H, 17-H), 4.50 (q, J=7.4 Hz, 1H, 18-H), 4.99 (s, 2H, 3a-H), 6.27 (s, 1H, 132-H), 8.58, 9.67, 10.22 (each s, each 1H, meso-H), 10.25 (s, 1H, 3a-CHO), 11.13 (s, 1H, 7-CHO); 13C NMR (CDCl3) δ: 203.0, 201.3, 173.5, 173.3, 161.1, 151.7, 149.0, 145.0, 136.4, 136.0, 131.2, 130.6, 129.6, 129.2, 128.3, 122.5, 140.0, 97.2, 93.1, 56.6, 51.6, 50.9, 39.1, 31.0, 30.3, 25.8, 23.4, 19.5, 17.4, 12.1, 11.2; IR (KBr) v: 3459 (N—H), 2958 (C—H), 1737, 1717 (C=O), 1662 (C=C), 1564 (chlorin skeleton), 1443, 1373, 1241, 1070, 908 cm-1; EI-MS m/z: 637.5 (M+H+). Anal. calcd for C36H36N4O7: C 67.91, H 5.70, N 8.80; found C 67.80, H 5.81, N 8.77.
3.11 12-甲酰基脱镁叶绿酸-d甲酯(12) 的合成
将200 mg LiOH溶解于3 mL水中, 在加入10 mL甲醇.搅拌下将该溶液倒入含有160 mg MPPa (1b, 0.292 mmol)的15 mL二氯甲烷中, 室温条件下避光搅拌4 h, 用乙酸将反应液的pH值调至3, 在加入20 mL水和20 mL二氯甲烷分层, 水相用二氯甲烷萃取(15 mL×3), 合并有机层并水洗, 用无水硫酸钠干燥.减压浓缩, 剩余物硅胶柱层析分离[洗脱剂: V(石油醚):V(乙酸乙酯)=4:1], 得到46 mg暗红色固体11(0.082 mmol, 28%).将其溶解于含有0.1 mL吡啶的15 mL四氢呋喃, 在0 ℃剧烈搅拌下, 将溶解在2 mL四氢呋喃中的24 mg OsO4 (0.094 mmol)慢慢滴加到反应液中, 保持温度并搅伴30 min.再于室温下搅拌反应2 h.加入20 mL在50%甲醇中饱和的NaHSO3溶液, 搅拌20 min, 过滤除去OsO3的红褐色沉淀.剧烈搅拌下向反应液中加入1 g硅胶, 然后将溶解于5 mL水中的200 mg NaIO4 (0.935 mmol)倒入反应体系, 颜色迅速由墨绿色变为铜色, 30 min后加入100 mL水和80 mL二氯甲烷淬灭反应, 过滤, 分出有机层, 干燥后用重氮甲烷甲基化.减压除尽溶剂, 剩余物经硅胶柱层析分离[洗脱剂: V(丙酮):V(甲苯)=1:10], 得到25 mg红色固体12 (0.043 mmol), 产率53%. m.p. 233~235 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 421 (11.30×104), 513 (7.91×103), 546 (3.05×104), 666 (2.71×104), 693 (4.97×104) nm; 1H NMR (400 MHz. CDCl3) δ: -0.43 (br s, 1H, NH), 0.58 (br s, 1H, NH), 1.58 (t, J=7.6 Hz, 3H, 8-CH3), 1.81 (d, J=7.2 Hz, 3H, 18-CH3), 1.87~2.08 (m, 1H, 17a+17b-H), 2.18~2.43 (m, 2H, 17a+17b-H), 2.54~2.78 (m, 1H, 17a+17b-H), 3.04, 3.61, 3.65 (each s, each 3H, CH3+OCH3), 3.45 (q, J=7.6 Hz, 2H, 8a-H), 4.16 (d, J=8.4 Hz, 1H, 17-H), 4.36 (q, J=7.2 Hz, 1H, 18-H), 5.03 (d, J=20.0 Hz, 1H, 132-H), 5.20 (d, J=20.0 Hz, 1H, 132-H), 8.49, 9.42, 9.68 (each s, each 1H, meso-H), 11.29 (s, 1H, 3-CHO), 11.33 (s, 1H, 12-CHO); 13C NMR (CDCl3) δ: 204.2, 204.0, 199.8, 174.5, 172.7, 170.6, 170.3, 146.0, 145.9, 142.5, 138.6, 137.4, 136.9, 133.3, 129.7, 129.1, 123.6, 105.6, 98.1, 62.5, 61.5, 56.2, 51.6, 51.2, 31.9, 31.4, 24.4, 24.2, 23.6, 23.4, 13.7, 13.0, 12.1; IR (KBr) v: 3449 (N—H), 2924 (C—H), 1742~1708 (C=O), 1686 (C=C), 1560 (chlorin skeleton), 1457, 1341, 1082, 1040, 971 cm-1. EI-MS m/z: 565.4 (M+H+). Anal. calcd for C33H32N4O5: C 70.20, H 5.71, N 9.92; found C 70.40, H 5.84, N 9.87.
3.12 N-羟乙基-12-甲酰基红紫素-18二酰亚胺甲酯(14) 的合成
将248 mg (0.409 mmol)化合物1a溶解于25 mL二氯甲烷中, 加入由氢氧化钾饱和的5 mL甲醇溶液, 室温搅拌1.5 h, 加入20 mL水, 搅拌下滴加浓盐酸调节pH至3.加入30 mL二氯甲烷分出有机层, 水相用二氯甲烷萃取(40 mL×2), 合并有机层, 水洗两次后用无水硫酸钠干燥, 减压除尽溶剂, 用少量2 mL二氯甲烷将固体混合物溶解, 移至含有1.5 mL羟乙胺的20 ml干燥的甲苯中, 搅拌回流8 h, 减压除去反应溶剂, 将所得粗产物13重新溶解在20 mL四氢呋喃中, 将反应体系温度降低至0 ℃, 加入氢氧化锂溶液(400 mg LiOH溶解在20 mL甲醇和5 mL水中), 室温搅拌6 h, 用25%的硫酸溶液调至pH值为2, 用CH2Cl2萃取反应液(15 mL×3), 合并有机层, 水洗后用无水硫酸钠干燥, 减压浓缩, 剩余物用硅胶柱层析[洗脱剂: V(石油醚):V(乙酸乙酯)=3:1]分离, 得73 mg黄绿色固体14 (0.115 mmol).产率28%. m.p. 225~228 ℃; UV-vis (CH2Cl2) λmax [ε/(L•mol-1•cm-1)]: 382 (5.23×104), 422 (1.03×103), 508 (9.22×102), 548 (5.15×103), 715 (9.02×103) nm; 1H NMR (400 MHz, CDCl3) δ: 0.73 (br s, 1H, NH), 0.80 (br s, 1H, NH), 1.54 (t, J=7.6 Hz, 3H, 8a-CH3), 1.62 (d, J=7.2 Hz, 3H, 18-CH3), 1.78~1.98 (m, 1H, 17a+17b-H), 2.27~2.46 (m, 2H, 17a+17b-H), 2.61~2.69 (m, 1H, 17a+17b-H), 2.91, 3.13, 3.54 (each s, each 3H, CH3+OCH3), 3.43 (q, J=7.6 Hz, 2H, 8a-H), 4.07~4.13 (m, 3H, CH2OH+18-H), 4.67 (t, J=5.0 Hz, 2H, NCH2), 5.03 (dd, J=8.9, 2.0 Hz, 1H, 18-H), 4.75 (dd, J=9.0, 3.0 Hz, 1H, 17-H), 6.09 (d, J=11. 6 Hz, 1H, cis-3b-H), 6.16 (d, J=17.8 Hz, 1H, trans-3b-H), 7.63 (dd, J=17.8, 11.6 Hz, 3a-H), 8.13, 8.76, 10.18 (each s, each 1H, meso-H), 11.73 (s, 1H, 12-CHO); 13C NMR (CDCl3)δ: 196.3, 173.8, 171.7, 159.6, 150.7, 148.9, 146.7, 144.8, 144.6, 139.6, 138.9, 135.6, 134.0, 132.6, 131.8, 131.7, 118.9, 118.0, 106.0, 104.9, 96.8, 51.8, 51.6, 49.4, 49.6, 30.9, 29.8, 29.7, 22.1, 20.5, 19.3, 16.9, 16.3, 15.9, 15.4, 11.4; IR (KBr) v: 3454 (N—H), 2956 (C—H), 1741, 1706 (C=O), 1604 (C=C), 1523 (chlo in skeleton), 1463, 1365, 1229, 1032, 989 cm-1; EI-MS m/z: 636.2 (M+H+). Anal. calcd for C36H37N5O6: C 68.02, H 5.87, N 11.02; found C 68.21, H 6.01, N 11.09.
3.13 N-甲酰乙基-12-甲酰基红紫素-18二酰亚胺甲酯(15) 的合成
以红紫素-18二酰亚胺醇14替代二氢卟吩醇7, 按照制备化合物8的方法合成.产率为50%; mp. 212~214 ℃; UV-vis (CH2Cl2)λmax [ε/(L•mol-1•cm-1)]: 433 (1.06×105), 452 (9.06×104), 576 (2.68×104), 632 (3.65×104), 718 (1.09×105) nm; 1H NMR (CDCl3) δ: 0.05 (br s, 1H, NH), 0.74 (br s, 1H, NH), 1.62 (t, J=7.6 Hz, 3H, 8a-CH3), 1.67 (d, J=7.2 Hz, 3H, 18-CH3), 1.85~1.95 (m, 1H, 17a+17bH), 1.98~2.09 (m, 1H, 17a+17b-H), 2.28~2.50 (m, 1H, 17a+17bH), 2.65~2.75 (m, 1H, 17a+17b-H), 3.52 (q, J=7.6 Hz, 2H, 8a-H), 2.99, 3.20, 3.61 (each s, each 3H, CH3+OCH3), 4.17 (q, J=7.2 Hz, 1H, 18-H), 5.06 (dd, J=9.0, 2.8 Hz, 1H, 18-H), 5.27 (s, 2H, N-CH2), 6.16(d, J=11.6, 1H, cis-3b-H), 6.24 (d, J=17.6, 1H, trans-3b-H), 7.70 (dd, J=17.6, 11.6 Hz, 1H, 3a-H), 8.20, 8.84, 10.27 (each s, each 1H, meso-H), 9.92 (s, 1H, NCH2CHO), 11.78 (s, 1H, 12-CHO); IR (KBr) v: 3450 (N—H), 2863 (C—H), 1736, 1699 (C=O), 1637 (C=C), 1556 (chlorin skeleton), 1402, 1266, 1094, 1027, 802 cm-1; EI-MS m/z: 634.3 (M+H+). Anal. calcd for C36H35N5O6: C 68.23, H 5.57, N 11.05; found C 68.16, H 5.49, N 11.26.
3.14 叶绿素二氢卟吩与蛋白质相互作用的测定
分别选择二氢卟吩化合物1a, 3, 11和13为待测化合物, 用四氢呋喃(THF, 优级纯)配制成1×10-4 mol• L-1的溶液, 暗室中常温保存; 牛血清白蛋白(BSA, 北京军区兽医防治中心生物工程研究室, 纯度>98%, 分子量68000), 加高纯水配置成1 mg•mL-1储备溶液, 置于1~4 ℃冰箱中保存.
移取1 mg•mL-1储备溶液的牛血清白蛋白0.2 mL与10 mL比色管中, 依次加入一定量1×10-4 mol•L-1叶绿素衍生物, 以蒸馏水定容至刻度, 在日立850型荧光光度计上以1 cm石英比色皿测定荧光强度, 狭缝宽(带通)为5 nm, λex为225 nm, λem为340 nm.
移取1×10-4 mol•L-1叶绿素衍生物0.6 mL于10 mL比色管中, 以蒸馏水定容至刻度, 在日立3400型UV-Vis分光光度计上进行吸收光测定.
辅助材料(Supporting Information)所有新化合物的1H NMR图谱.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
-
-
[1]
(a) Nyman, E. S.; Hynninen, P. H. J. Photochem. Photobiol. A 2004, 73, 1.
(b) Pavlov, V. Y.; Ponomarev, G. V. Chem. Heterocycl. Compd. 2004, 40, 393.
(c) Wang, J.-J. Chin. J. Org. Chem. 2005, 25, 1353(in Chinese). (王进军, 有机化学, 2005, 25, 1353.)
(d) Goswami, L. N.; Ethirajan, M.; Dobhal, M. P.; Zhang, M.; Missert, J. R.; Shibata, M.; Kadish, K. M.; Pandey, R. K. J. Org. Chem. 2009, 73, 568 -
[2]
(a) Okamoto, Y.; Tamiaki, H. J. Photochem. Photobiol. A 2011, 209, 250.
(b) Li, J.; Liu, Y.; Xu, X.-S.; Li, Y.-L.; Zhang, S.-G.; Yoon, I.; Shim, Y.K.; Wang, J.-J.; Yin, J.-G. Org. Biomol. Chem. 2015, 13, 1992.
(c) Li, Y.-L.; Li, J.-Z.; Zhang, S.-G.; Wang, J.-J. Chin. J. Org. Chem. 2016, 36, 562(in Chinese). (李彦龙, 李家柱, 张善国, 王进军, 有机化学, 2016, 36, 562.)
(d) Li, J.; He, N.; Liu, Y.; Zhang, Z.; Zhang, X.; Han, X.; Gai, Y.; Liu, Y.; Yin, J.; Wang, J. Dyes Pigm. 2017, 146, 189. -
[3]
(a) Li, J.-Z.; Wang, J.-J.; Yoon, I.; Cui, B.-C.; Shim, Y. K. Bioorg. Med. Chem. Lett. 2012, 22, 1846.
(b) Liu, H.-Y.; Zhu, G.-H.; Liu, Y.-Y.; Jin, Y.-X.; Qi, C.-X.; Wang, J.-J. Chin. J. Org. Chem. 2015, 35, 1320(in Chinese). (刘红瑶, 朱国华, 刘冉冉, 金英学, 祁彩霞, 王进军, 有机化学, 2015, 35, 1320. -
[4]
Ali, H.; Vanlier, J. E. In Handbook of Porphyrin Science, Vol. 4, Eds.:Kadish, K. M.; Smith, K. M.; Guilard, R., World Scientific Publishing Company, Singapore, 2010, pp. 1~119.
-
[5]
Ethirajan, M.; Patel, N. J.; Pandey, R. K. In Handbook of Porphyrin Science, Vol. 4, Eds.:Kadish, K. M.; Smith, K. M.; Guilard, R., World Scientific Publishing Company, Singapore, 2010, pp. 249~323.
-
[6]
Wang, X. F.; Tamiaki, H. Energy Environ. Sci. 2010, 3, 94 doi: 10.1039/B918464C
-
[7]
(a) Yin, J.-G.; Wu, X.-R.; Liu, C.-L.; Zhao, L.-L., Jin, Y.-X.; Wang, J.-J. Chin. J. Org. Chem. 2011, 31, 1870(in Chinese). (殷军港, 邬旭然, 刘春林, 赵丽丽, 金英学, 王进军, 有机化学, 2011, 31, 1870.)
(b) Yin, J.-G.; Wang, Z.; Yang, Z.; Liu, C.; Zhao, L.-L.; Wang, J.-J. Chin. J. Org. Chem. 2012, 32, 360(in Chinese). (殷军港, 王振, 杨泽, 刘超, 赵丽丽, 金英学, 王进军, 有机化学, 2012, 32, 360. -
[8]
(a) Wang, J.-J.; Shim, Y. K.; Jiang, G.-J.; Imafuku, K. J. Heterocycl. Chem. 2004, 41, 29.
(b) Wang, J.-J.; Li, J.-Z.; Wu, X.-R.; Shim, Y. K. Chin. J. Chem. 2006, 24, 933. -
[9]
(a) Wang, L.-M.; Wang, Z.; Yang, Z.; Jin, Y.-X.; Wang, J.-J. Chin. J. Org. Chem. 2012, 32, 2154(in Chinese). (王鲁敏, 王振, 杨泽, 金英学, 王进军, 有机化学, 2012, 32, 2154.)
(b) Ji, J.-Y.; Xia, S.-W.; Liu, Y.; Yin, J.-G.; Qi, C.-X.; Wang, J.-J. Chin. J. Org. Chem. 2014, 34, 1138(in Chinese). (纪建业, 夏尚文, 刘洋, 殷军港, 祁彩霞, 王进军, 有机化学, 2014, 34, 1138. -
[10]
刘洋, 硕士论文, 烟台大学, 烟台, 2014.Liu, Y. M.S. Thesis, Yantai University, Yantai, 2014(in Chinese).
-
[11]
(a) Hu, X.-L.; Cui, S.-Y.; Liu, J.-Q. Spectrochim. Acta A 2010, 77, 548.
(b) Jia, F.-G.; Wang, W.-W.; Wang, J.; Yin, J.-G.; Liu, Y.-M.; Liu, Z.-B. Anal. Methods 2012, 4, 449. -
[12]
(a) Smith, K. M.; Gogg, D. A.; Simpson, D. J. J. Am. Chem. Soc. 1985, 107, 4946.
(b) Schenk, J.; Daessler, H. G. Pharmazie 1969, 24, 419.
-
[1]
-

图式1 单醛基取代叶绿素-a衍生物的合成
Scheme 1 Synthesis of mono-aldehyde group-substituted chlorophyll a derivatives
Ragents and conditions: (a) AcOH/110 ℃; (b) KOH/MeOH/Pyr/reflux; (c) Ni(OAc)2/DMF/POCl3; (d) H2SO4/CH2Cl2; (e) KOH/MeOH/Zn(OAc)2; (f) (CH3)2NCH=CHCHO/POCl3; (g) H2/Pd-C/HCl/AcOH/H3PO4; (h) TPAP/N-MMO

图式2 双醛基取代叶绿素-a衍生物的合成
Scheme 2 Synthesis of di-aldehyde group-substituted chlorophyll a derivatives
Reagents and conditions: (a) TI(NO3)•3H2O; (b) 88% HCO2H; (c) LiOH/MeOH/THF; (d) OsO4/NaIO4; (e) KOH/MeOH/NH2C2H4OH/Py; (f) TPAP/N-MMO.

图 1 焦脱镁叶绿酸的色基结构互变
Figure 1 Interconversion of the chromophore structure in pyropheophorbide

图 2 叶绿素二氢卟吩的20-位亲电取代反应
Figure 2 The electrophilic substitution reactions of chlorophyllous chlorins at 20-position

图 3 二氢卟吩11 (a)与BSA(b)的同步荧光图谱
Figure 3 Synchronous fluorescence spectrum of 11 (a) and BSA (b)
c11=1.00×10−6 mol•L−1; cBSA=1.47×10−5 mol•L−1; λex=225 nm, λem=340 nm
表 1 lg(F0-F)/F与lg[Q]的方程、平衡常数K0和结合数n
Table 1. Equation, K0 and n of lg(F0-F)/F vs. lg[Q]
叶绿素二氢卟吩 T/℃ lg(F0-F)/F vs. lg[Q] equation K0/(L·mol-1) n 1a 25 y=1.0457x+5.009 (R2=0.9954) 1.02×105 1.0457 37 y=1.1330x+5.6234 (R2=0.9974) 4.20×105 1.1330 3 35 y=1.2677x+5.9916 (R2=0.9966) 5.58×105 1.2677 37 y=0.9861x+4.8179 (R2=0.9959) 6.58×104 0.9861 11 25 y=1.2592x+6.5379 (R2=0.9977) 3.45×106 1.2592 37 y=1.2914x+6.5952 (R2=0.9902) 3.94×106 1.2914 13 25 y=1.1309x+5.5556 (R2=0.9954) 3.60×105 1.1309 37 y=0.9908x+4.7324 (R2=0.9902) 5.40×104 0.9908  下载: 导出CSV
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 39
- 文章访问数: 2793
- HTML全文浏览量: 352
 下载:
下载:
