Citation:
Ling Fang, Sha Wang, Shun Lu, Fengjun Yin, Yujie Dai, Lin Chang, Hong Liu. Efficient electroreduction of nitrate via enriched active phases on copper-cobalt oxides[J]. Chinese Chemical Letters,
2024, 35(4): 108864.
doi:
10.1016/j.cclet.2023.108864
Efficient electroreduction of nitrate via enriched active phases on copper-cobalt oxides
English
Efficient electroreduction of nitrate via enriched active phases on copper-cobalt oxides
Chongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, Chongqing 400714, China
b.
University of Chinese Academy of Sciences, Beijing 100049, China
* Corresponding author. E-mail address: liuhong@cigit.ac.cn (H. Liu). 1 These authors contributed equally to this work.
Received Date:
03 April 2023 Accepted Date:
27 July 2023 Revised Date:
05 July 2023 Available Online:
15 April 2024
Abstract:
Electrochemical conversion of nitrate (NO3−) to ammonia (NH3) can target two birds with one stone well, in NO3−-containing sewage remediation and sustainable NH3 production. However, single metal-based catalysts are difficult to drive high-efficient NO3− removal due to the multi-electron transfer steps. Herein, we present a tandem catalyst with simple structure, Cu-Co binary metal oxides (Cu-Co-O), by engineering intermediate phases as catalytic active species for NO3− conversion. Electrochemical evaluation, X-ray photoelectron spectroscopy, and in situ Raman spectra together suggest that the newly-generated Cu-based phases was prone to NO3− to NO2− conversion, then NO2− was reduced to NH3 on Co-based species. At an applied potential of −1.1 V vs. saturated calomel electrode, the Cu-Co-O catalyst achieved NO3−-N removal of 90% and NH3 faradaic efficiency of 81% for 120 min in 100 mL of 50 mg/L NO3−-N, consuming only 0.69 kWh/mol in a two-electrode system. This study provides a facile and efficient engineering strategy for developing high-performance catalysts for electrocatalytic nitrate conversion.
Nitrate (NO3−) pollution has become one of the major concerns in water management [1]. High-strength NO3− wastewater that was mainly induced by anthropogenic activities such as fertilizer and industrial applications poses a potential threat to human health [2,3]. Electrocatalytic NO3− reduction reaction (NO3RR) highlights its advantages particularly for industrial wastewater (e.g., wastewater from explosives and photovoltaic industries), where the NO3− content is high and bacterial growth is unviable [4,5]. Instead of converting NO3− into N2 for purification purposes, the acquisition of ammonia (NH3) from NO3− provides a valuable pathway for the production of NH3, an essential chemical and an alternative carbon-free energy carrier [6–8]. The separation of NH3 can be efficiently achieved using extraction technology or regenerated resins, while the electricity required for this process can be generated from clean and renewable sources such as solar and wind power [9,10]. Therefore, NO3RR may provide an opportunity for the NH3 production industry and remediation of NO3−-containing wastewater.
Cu-based catalysts have been widely explored for the NO3RR due to its high electrocatalytic reduction kinetics of NO3− to nitrite (NO2−), which is the rate-limiting step of the NO3RR to NH3 [11,12]. However, the intermediates NO2− will accumulate on the pure Cu electrocatalyst surface thereby leading to its rapid deactivation and increased energy consumption [13]. Substantial efforts such as alloying Cu with transition metals (e.g., Pt, Pd, and Ni) or formation of compounds with molecular solids have been made to overcome these limitations [14,15]. In particular, Cu-based oxide (CuO) was transformed into hybrids (Cu/Cu2O) in situ in NO3RR, which enhances NO3− removal and NH3 generation [16,17]. However, the conversion of NO3− into NH3 involves an eight-electrons transfer and multiple reaction intermediates. On the single metal oxide catalyst surface, optimizing the binding strength of one reactant would typically take the other steps away from their optima [18–20]. In this case, simultaneous acceleration of sequential reduction of oxygen-containing intermediates (e.g., *NO2− and *NO) and hydrogenation of nitrogen-containing intermediates (e.g., *NH and *NH2) remains challenging.
A tandem electrocatalyst contains distinct active sites proceeding the individual reaction steps, which can couple and mediate the consecutive steps to effectively produce the desired products [21–23]. Transition metal oxides undergo potential-dependent phase formation during cathodic electrocatalysis, resulting in the coexistence of multiple species such as metallic, oxide, and hydroxide [24–26]. It presents an opportunity for optimizing the electrocatalytic performance if these freshly-generated species could be combined for tandem NO3− to NH3 conversion. As mentioned above, Cu-based species suffer from the adsorption of NO3−-reduction intermediates such as NO2− to block the electrode surface and slow down subsequent deoxidation and hydrogenation process [27,28]. Fortunately, Co-based oxides have been reported to exhibit satisfactory activity towards NO3− to NH3 and specific NO2− reduction to NH3 [29,30]. Therefore, we hypothesize that combining the advantages of Cu-based catalyst with high reduction kinetics of NO3− to NO2− and Co-based catalyst with favorable conversion towards NO2− to NH3 would accelerate sequential NO3− to NH3 with low overpotential.
In this work, a series of Cu-Co binary metal oxides (Cu-Co-O) with different Cu and Co content were prepared combining facile co-precipitation and calcination method. Interestingly, multiple species were formed in situ on the Cu-Co-O surface during the NO3RR, which acted as tandem active species for NO3− to NH3 conversion. Electrocatalytic measurements, X-ray photoelectron spectroscopy (XPS), in situ Raman spectra, and in situ differential electrochemical mass spectrometry (DEMS) revealed that Cu-based species are favorable for NO3− to NO2− conversion, while Co-based phases tend to catalyze NO2− reduction to NH3. Consequently, Cu-Co-O catalysts demonstrated higher NO3− removal and NH3 generation compared to single metal oxide (CuO or Co3O4) at low overpotentials. This study presents a general strategy for designing high-performance tandem catalysts for multi-step electrochemical reactions.
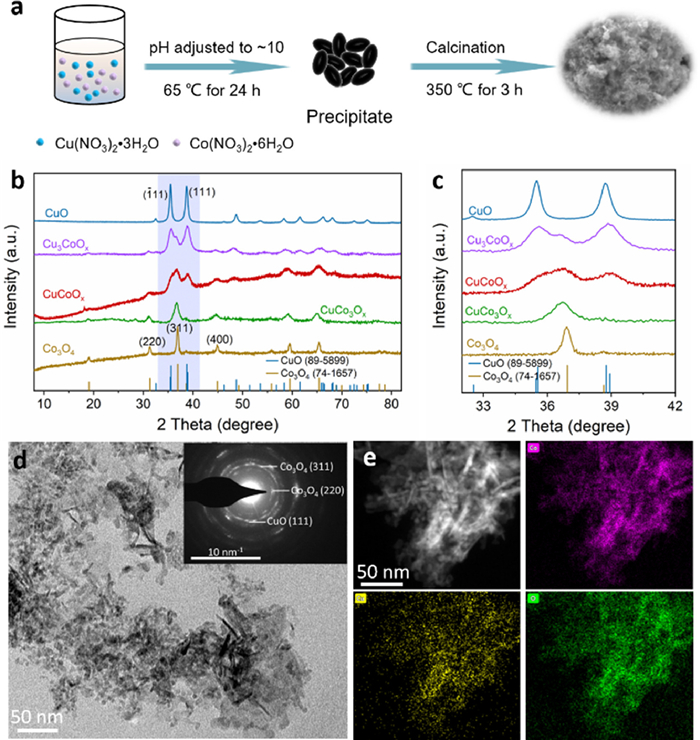
The Cu-Co-O catalysts were synthesized via a facile co-precipitation method coupled with subsequent calcination (Fig. 1a). By carefully adjusting the molar ratio of Cu to Co in the co-precipitation process, the Cu-Co-O with tunable element components could be acquired (Table S1 in Supporting information). Powder X-ray diffraction (XRD) patterns (Fig. 1b) show that main peaks located at 35.6° and 38.8° were assigned to the (111) and (111) crystal planes of CuO (JCPDS No. 89–5899), and the main peaks at 31.3°, 36.9° and 44.9° were well indexed to the (220), (311) and (400) of Co3O4 (JCPDS No. 74–1657). Comparing the diffraction peaks of these obtained samples, we found that the relative intensity of CuO signals were gradually weakened with the introduction of Co element. The observed peak shifts of the samples (Fig. 1c) synthesized using mixed metal sources, as compared to Co3O4 and CuO, suggested the doping of Co3O4 with Cu or CuO with Co that caused the contraction or expansion of the crystal lattice, respectively [31,32]. Furthermore, the diffraction signals of both CuO and Co3O4 were evident in Cu3CoOx, CuCoOx, and CuCo3Ox, indicating the composite nature of these catalysts. In addition, XPS analysis of the pristine catalyst revealed no nitrogen signals, suggesting that the use of metal precursor containing NO3− had no effect on the accurate quantification of NO3− in water (Fig. S2 in Supporting information).
Figure 1
Figure 1.
(a) Synthesis schematic of the Cu-Co-O catalysts. (b) XRD patterns of the Cu-Co oxides. (c) Enlarged XRD patterns corresponding to the blue area of (b). (d) TEM image and inset is the SAED image of CuCoOx. (e) Color elemental mapping of Co (purple), Cu (yellow), and O (green), respectively.
The morphological characteristics of the prepared CuCoOx were uncovered by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1d and Figs. S3a-d (Supporting information), CuCoOx exhibited assembled nanosheets with a rough surface and the addition of Na2CO3 could modify the particle size and morphology of CuCoOx (Fig. S3e in Supporting information). The TEM images, coupled with selected area electron diffraction (SAED), display that the nanosheets were mainly composed of nanocrystals and featured a polycrystalline structure (Fig. 1d). The corresponding energy dispersive X-Ray spectroscopy (EDX) mapping reveal strong and evenly distributed signals of Co, Cu, and O throughout the entire selected area (Fig. 1e).
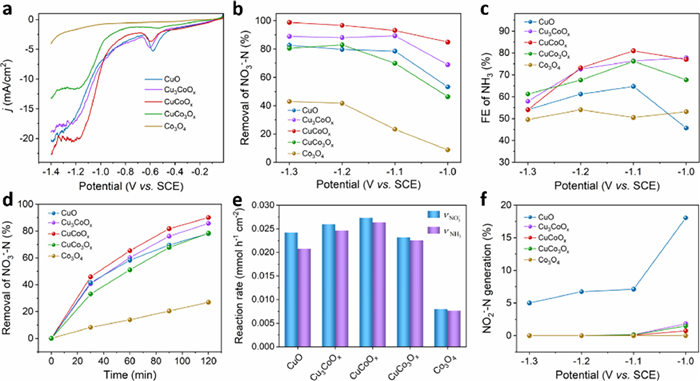
Linear sweep voltammetry (LSV) and two-hour electrolysis were conducted to assess the performance of the obtained catalysts. Fig. 2a shows LSV curves in the presence of NO3−. As observed, CuCoOx catalyst displayed increased current density (j) compared with CuO and Co3O4. Moreover, CuCoOx showed a clear increase of the reductive current after adding NO3−, implying a catalytic response towards the NO3RR (Fig. S4 in Supporting information). CuCoOx demonstrated the highest NO3− removal over the entire potential range from −1.0 V to −1.3 V, with NO3−-N removal percentage exceeding 90% at −1.1 V (Fig. 2b). Moreover, the catalytic current increased as the electrolysis potential became more negative, and CuCoOx achieved a maximum FE of 81% for NH3 at −1.1 V (Fig. 2c and Fig. S5 in Supporting information). At this applied potential, CuO exhibited low FE for NH3, while Co3O4 showed weak NO3RR activity. Notably, in some reported literature, single Cu- or Co- based catalysts required more negative potentials to achieve similar percentages of NO3− removal (Table S2 in Supporting information) [33,34], indicating that the synergy of Cu and Co sites in CuCoOx may reduce the overpotential of NO3RR.
Figure 2
Figure 2.
(a) LSV curves of the prepared Cu-Co oxides. (b) Removal percentage of NO3−-N on Cu-Co oxides at different potentials. (c) FE of NH3 on Cu-Co oxides at different potentials. (d) NO3−-N removal. (e) vNO3− and vNH3 (for the NO3RR on Cu-Co oxides at −1.1 V. (f) NO2−-N generation during NO3RR on the prepared catalysts at different potentials. Figs. b, c, e and f are from 120 min electrolysis in 100 mL solution with 50 mg/L NO3−-N.
Furthermore, we analyzed the product distribution catalyzed by these catalysts through continuous electrolysis measurement at a fixed potential. Over the potential range of −1.0 V to −1.3 V, the NO3−-N removal gradually increased, while the FE of NH3 exhibited a maximum value at −1.1 V for almost all of the prepared samples (Figs. 2b and c). Thus, we selected −1.1 V for potentiostatic electrolysis. As shown in Fig. 2d and Fig. S6 (Supporting information), NO3−-N was removed while NH4+-N kept increasing with the prolonging of reaction time. CuCoOx exhibited the best NO3−-N removal (90%) and NH4+-N generation (87%). The values of vNO3− on CuCoOx (0.027 mmol h−1 cm−2, 0.026 mmol h−1 cm−2) at 120 min were higher than those on CuO (0.024 mmol h−1 cm−2, 0.021 mmol h−1 cm−2) and Co3O4 (0.008 mmol h−1 cm−2, 0.007 mmol h−1 cm−2) (Fig. 2e). Notably, above 6% of NO2−-N was generated on CuO during the 120 min reaction, but there was no evident NO2−-N accumulation after the introduction of Co in CuO (Fig. S7 in Supporting information). Since NO2− is an even more dangerous pollutant than NO3−, it is desirable to keep its content as low as possible [35]. Additionally, NO2−-N was detected in the case of Cu-Co-O electrodes only when the applied potential potentials were more positive than −1.1 V (Fig. 2f). These results imply that Cu-based species can convert NO3− to NO2−, and Co-based species might further reduce NO2− to NH3.
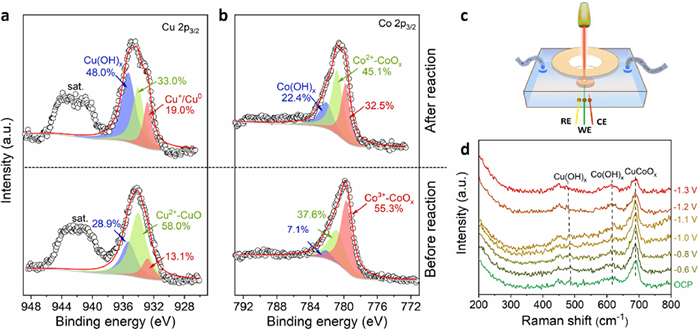
XRD and XPS were conducted to identify the compositions of the Co3O4, CuO, and CuCoOx catalysts. Fig. S8 (Supporting information) is the XRD profile of the three catalysts after 120 min of NO3RR. The carbon signals appeared because the prepared catalyst powders were loaded on the carbon paper. After NO3RR, CuO showed obvious Cu0 signals. It was difficult to observe the diffraction peak indexed to the metal hydroxides or reductive metal oxides due to the extremely low content or poor crystallization of the generated species in the samples. XPS was further carried out to analyze the species on the catalyst surface. High-resolution Co 2p and Cu 2p spectra displayed spin orbit splitting into 2p1/2 and 2p3/2 segments, and both segments qualitatively contained the same chemical valence information [36,37]. Therefore, only the higher-intensity Co 2p3/2 and Cu 2p3/2 bands were fitted in the obtained spectra. Cu 2p spectra of CuCoOx showed increased signals associated with reductive phases (Cu(OH)x and Cu+/Cu0 based species) after the NO3RR (Fig. 3a). It should be noted that Cu 2p spectra of CuCoOx after NO3RR revealed the presence of observable satellite features at 943.0 eV, whereas the satellite signals on CuO after the reaction were almost undetectable (Fig. 3a and Fig. S10a in Supporting information). These results confirm that Cu species predominantly existed in a positive-valence state rather than a zero-valence state in CuCoOx after reaction, and the opposite was true for CuO after the reaction [38,39]. Cu LMM spectra are more useful in determining chemical states of Cu. As shown in Fig. S9a (Supporting information), Cu LMM indicated the presence of multiple Cu-based phases, including Cu0 (566.8 eV), CuO (568.7 eV), Cu2O (569.8 eV), and Cu(OH)x (571.0 eV) in CuCoOx after the NO3RR, while the pristine CuCoOx mainly contained CuO phase. Compared to CuO (Fig. S10b in Supporting information), CuCoOx showed higher content of Cu(OH)x phase, implying that the compositions in CuCoOx were impacted by the coexistent Co-based phases. Moreover, the content of Co2+-dominated CoO (780.8 eV) and Co(OH)x (782.1 eV) in CuCoOx significantly increased after NO3RR, which could be also observed in Co3O4 before and after reaction (Fig. 3b and Fig. S11a in Supporting information). The composition changes can be further confirmed by the O 1s spectra (Figs. S9b, S10c and S11b in Supporting information). Oxygen species with binding energies below 531 eV are characteristic metal-oxygen in metal oxides, while signals at 531–532 eV correspond to metal hydroxides. Furthermore, the peak at the higher values above 532 eV is associated with oxygen vacancies [40,41]. As expected, the content of metal hydroxides and oxygen vacancies increased markedly, while the metal-oxide phases decreased for all three catalysts after the NO3RR.
Figure 3
Figure 3.
(a) Cu 2p and (b) Co 2p XPS spectra of CuCoOx before and after 120 min of the NO3RR. (c) Schematic diagram of the in situ Raman experiment. (d) In situ Raman spectra of CuCoOx at different applied potentials in flowing electrolytes containing 50 mg/L NO3−-N and 0.5 mol/L Na2SO4.
To gain more insight into the phase evolution of the three catalysts at different applied potentials, we interrogated the catalyst surface using in situ Raman spectroscopy (Fig. 3c). The Co3O4 and CuO powder exhibited their characteristic Raman bands at 190/482/521/690 cm−1 and 293/340/628 cm−1 (Fig. S12 in Supporting information), respectively [25,42]. Note that the spectral response of CuCoOx was not a direct combination of the spectra of Co3O4 and CuO, but displayed significant shoulder peaks, implying the interactions between Cu and Co element within the CuCoOx catalyst. We then acquired Raman spectra on the prepared three electrodes (CuO, Co3O4, and CuCoOx) at the applied potentials from the open-circuit potential (OCP) to −1.3 V vs. SCE (Fig. 3d and Fig. S13 in Supporting information). Under negative potentials, the peaks associated with CuO gradually disappeared, which could be attributed to the conversion of CuO to related reduced Cu-based species but difficult to distinguish from background noise across the entire series (Fig. S13a). On the Co3O4 electrode, the typical Raman peaks related to Co3O4 were attenuated with reducing potential while the signals about Co(OH)x were enhanced after the potential of −1.0 V (Fig. S13b). In the case of CuCoOx, the Raman signals associated with CuCoOx gradually weakened, while bands corresponding to Cu(OH)x and Co(OH)x became increasingly evident at potentials below −0.8 V (Fig. 3d).
These findings indicate that new species, such as reductive metal oxides and metal hydroxides, were generated on the catalyst surface during NO3RR, which may provide enriched active phases for enhanced NO3RR. Based on the above results, CuCoOx was a combination of CuO and Co3O4 in phase compositions. Therefore, it is reasonable to speculate that CuCoOx catalyst exhibits tandem reactivity involving Cu-based and Co-based species. The aforementioned electrochemical results indicate that NO2− accumulated when CuO was used as catalyst, but not when Co3O4 was used. Accordingly, the sequential acceleration of the NO3− to NO2− on CuO and NO2− to NH3 on Co3O4 would lead to fast NO3− to NH3 conversion with high NH3 selectivity at low overpotentials.
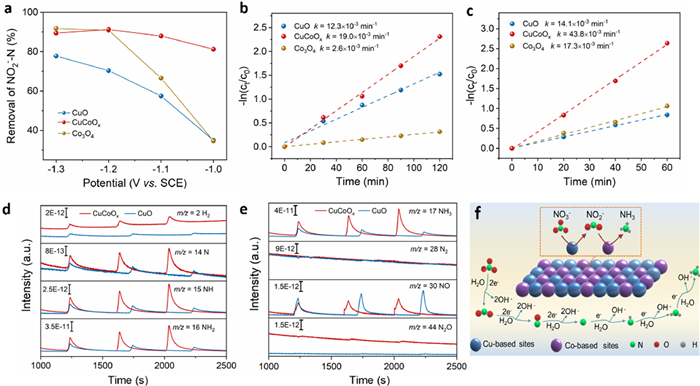
To confirm this hypothesis, we further evaluated NO2−-N removal performance of CuO, CuCoOx, and Co3O4 in a divided cell reactor. At −1.0 and −1.1 V, CuCoOx exhibited the highest NO2−-N removal compared to Co3O4 and CuO, and Co3O4 showed better NO2−-N removal than CuO (Fig. 4a). Moreover, CuCoOx inherited the advantages of Co3O4 and reached a maximum FE of 93.3% for NH3 at −1.1 V, which was higher than that of Co3O4 (81%) and CuO (66.5%) (Fig. S14a in Supporting information). CuCoOx also displayed higher NO2−-N removal at −1.1 V at regular intervals (Fig. S14b in Supporting information). Furthermore, we evaluated the rate constants of the catalysts for NO3− and NO2− removal. As shown in Figs. 4b and c, CuO exhibited a larger rate constant (12.3 × 10−3 min−1) for NO3− removal than Co3O4 (2.6 × 10−3 min−1), but a smaller rate constant (14.1 × 10−3 min−1) for NO2− removal than Co3O4 (17.3 × 10−3 min−1). It suggests that CuO was more effective in reducing NO3− while Co3O4 reduced NO2− faster than CuO.
Figure 4
Figure 4.
(a) Removal of NO2−-N at different potentials (100 mL solution with 15 mg/L NO2−-N, 60 min treatment). Plots of −ln(ct/c0) versus time for (b) NO3−-N removal and (c) NO2−-N removal on CuO, CuCoOx, and Co3O4 at −1.1 V. The values in the figure are the corresponding rate constant (k). (d, e) DEMS measurements of the NO3RR on CuCoOx and CuO (three cycles under the potential of −1.1 V). (f) Proposed mechanism of NO3RR on Cu-Co-O catalysts.
Furthermore, the molecular intermediate and product over CuO, CuCoOx, and Co3O4 were observed by in situ DEMS (Fig. S15 in Supporting information) [43]. As shown in Figs. 4d and e, the signals of NO, N, NH, NH2, NH3, and H2 were detected on the CuCoOx and CuO. The strong m/z 17 of NH3 signal and negligible m/z 28 of N2 signal confirmed the high NH3 selectivity of CuCoOx. Notably, H2 was not a preferred product on CuCoOx while Co3O4 yielded more H2 compared with CuCoOx and CuO (Fig. S16 in Supporting information). In general, NH3 as a NO3−-reduction product is favored in a potential region close to the hydrogen evolution reaction (HER), where NH3 would be formed by the reaction between adsorbed hydrogen (H*) and adsorbed NO3− [44,45]. Therefore, it can be inferred that Cu-based species reduce NO3− to N-containing intermediates, which can then be utilized by Co-based species to generate more deeply hydrogenated intermediates (e.g., NH, NH2 and NH3) and suppress competitive HER as well.
Cu-Co-O catalysts effectively enhance catalytic activity through the following mechanism. The NO3RR on Cu-Co-O catalysts to produce NH3 (NO3− + 6H2O + 8e− → NH3 + 9OH−) involves several elementary reactions: NO3− is initially adsorbed on the catalyst surface, followed by a series of deoxidation processes (*NO3− → *NO2− → *NO → *N) and sequential hydrogenation of *N (*N → *NH → *NH2 → *NH3). Under the reduction potential of the NO3RR, electrochemically driven phase generation occurs on the Cu-Co-O surface, resulting in the formation of hybrid M(OH)x/MOx phases that act as cooperative active sites for the multi-electron NO3RR. Specifically, Cu-based species as active sites preferentially adsorb NO3− and reduce it to NO2−, which is then sequentially reduced to NH3 on nearby Co-based sites. More importantly, the implementation of the tandem system makes it possible to lower the reaction overpotential while suppressing hydrogen evolution reaction, leading to highly efficient NO3− to NH3 conversion (Fig. 4f).
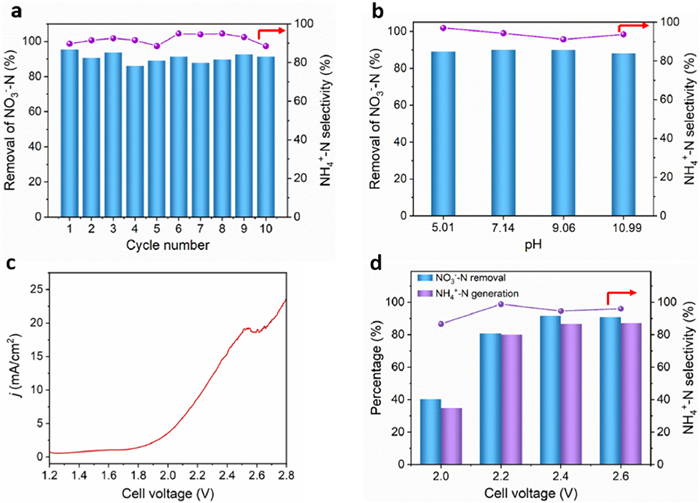
The long-term stability is another key parameter for electrocatalyst evaluations. Comparing obvious attenuation of the NO3−-N removal on CuO catalyst, CuCoOx showed insignificant variations for NO3−-N removal after 10 cycles (Fig. 5a and Fig. S17a in Supporting information). Moreover, the NH4+-N selectivity of CuCoOx remains ~90% after 10 cycles. In addition, the pristine assembled nanosheets morphology of CuCoOx is maintained after the cycling test, which further corroborated its favorable durability (Figs. S18a and b in Supporting information). The energy dispersive spectroscopy (EDS) mapping images of the CuCoOx after cycling test indicated the uniform distribution of Cu, Co, and O elements (Fig. S18c in Supporting information). During NO3RR, the pH of the electrolyte gradually became alkaline [46]. Previous studies have reported that single Cu catalyst gradually deactivated due to the formation of amorphous Cu resulting from the decomposition of CuH or CuH2, which inhibited electrochemical performance in alkaline solution [35]. This also explains the reason for the inferior stability of CuO, as above mentioned. Furthermore, we investigated the role of pH played on CuCoOx catalyst for the NO3−-N removal and NH4+-N selectivity. As shown in Fig. 5b and Fig. S17b (Supporting information), after 120 min treatment, NO3−-N removal and NH4+-N selectivity reached 90% or higher at different pH levels, implying that the CuCoOx catalyst prepared in this study had a broad working pH range. It is speculated that the Cu-Co compound could prevent the formation of amorphous Cu and promoted the presence of Cu2+/Cu+-based species, as demonstrated in XPS results. These Cu2+/Cu+-based species in CuCoOx served as active sites for achieving long-term reaction towards NO3RR. Furthermore, we investigated the impact of Cl− and CO32− on the NO3RR performance. The concentration of the impurities was set based on the NO3− concentration of 3.57 mmol/L. Our results demonstrated that the presence of Cl− and CO32− had no significant effect on the NO3−-N removal or the selectivity of NH4+-N (Fig. S17c in Supporting information).
Figure 5
Figure 5.
(a) The consecutive cycling test at −1.1 V on CuCoOx electrode. (b) The percentage of NO3−-N removal and NH4+-N selectivity on CuCoOx at different pH for 120 min treatment. (c) LSV curves of NO3RR on CuCoOx||Ir-Ru/Ti with a two-electrode configuration. (d) The percentage of NO3−-N removal and NH4+-N generation (left axis) and NH4+-N selectivity (right axis) on CuCoOx||Ir-Ru/Ti at different cell voltage.
In order to evaluate the potential practical applications of CuCoOx, we used a two-electrode system with CuCoOx cathode and Ir-Ru/Ti anode to investigate the performance of NO3RR. The polarization curve (Fig. 5c) revealed an increased j at the applied potential of 2.0 V. Potentiostatic electrolysis showed that the NO3−-N removal and NH4+-N selectivity reached > 90% at a cell voltage of 2.4 V after 120 min treatment (Fig. 5d and Fig. S19 in Supporting information). Accordingly, the energy consumption was calculated to be 0.69 kWh/mol, which was comparable to the FeNi/graphitized mesoporous carbon/Ni foam [47] and Pd-based TiO2 cathode [48].
In summary, we demonstrated that Cu-Co oxides exhibited highly tandem reactivity towards NO3− reduction. During NO3RR, electrochemical reduction induced phase reconstruction of Cu-Co oxides to generate more Cu- and Co-based phases as catalytic active species. In this tandem system, Cu-based species preferred to reducing NO3− to NO2−, while the generated NO2− intermediates would further be converted to NH3 on Co-based species. Benefiting from the favorable Cu- and Co-based partners, Cu-Co oxide electrode reached over 90% of nitrate removal and ammonia selectivity, which was superior than those of single metal oxides (CuO and Co3O4). Moreover, the good stability and low energy consumption (0.69 kWh/mol) of Cu-Co oxides were comparable to most of the NO3RR catalysts. These findings indicate a universal strategy for enhancing NO3− conversion into value-added NH3 with the efficient tandem electrocatalysts, which can also be extended to other multi-step electrocatalytic reaction.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Nos. 52131003 and 42007180), Special Research Assistant Program of Chinese Academy of Science, Natural Science Foundation of Chongqing (No. cstc2020jcyj-msxmX0775), Scientific Research Instrument Development Project of Chinese Academy of Sciences (No. YJKYYQ20200044), Outstanding Scientist of Chongqing Talent Program (No. CQYC20210101288).
Supplementary material
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108864
[1]
M. Duca, M.T.M. Koper, Energy Environ. Sci. 5 (2012) 9726–9742. doi: 10.1039/c2ee23062c
[2]
S. Leaković, I. Mijatović, Š. Cerjan-Stefanović, et al., Water Res. 34 (2000) 185–190. doi: 10.1016/S0043-1354(99)00122-0
[3]
Y. Zeng, C. Priest, G. Wang, et al., Small Methods 4 (2020) 2000672. doi: 10.1002/smtd.202000672
X.T. Chen, T. Zhang, M. Kan, et al., Environ. Sci. Technol. 54 (2020) 13344–13353. doi: 10.1021/acs.est.0c05631
[48]
P. Gayen, J. Spataro, S. Avasarala, et al., Environ. Sci. Technol. 52 (2018) 9370–9379. doi: 10.1021/acs.est.8b03038
Figure 1
(a) Synthesis schematic of the Cu-Co-O catalysts. (b) XRD patterns of the Cu-Co oxides. (c) Enlarged XRD patterns corresponding to the blue area of (b). (d) TEM image and inset is the SAED image of CuCoOx. (e) Color elemental mapping of Co (purple), Cu (yellow), and O (green), respectively.
Figure 2
(a) LSV curves of the prepared Cu-Co oxides. (b) Removal percentage of NO3−-N on Cu-Co oxides at different potentials. (c) FE of NH3 on Cu-Co oxides at different potentials. (d) NO3−-N removal. (e) vNO3− and vNH3 (for the NO3RR on Cu-Co oxides at −1.1 V. (f) NO2−-N generation during NO3RR on the prepared catalysts at different potentials. Figs. b, c, e and f are from 120 min electrolysis in 100 mL solution with 50 mg/L NO3−-N.
Figure 3
(a) Cu 2p and (b) Co 2p XPS spectra of CuCoOx before and after 120 min of the NO3RR. (c) Schematic diagram of the in situ Raman experiment. (d) In situ Raman spectra of CuCoOx at different applied potentials in flowing electrolytes containing 50 mg/L NO3−-N and 0.5 mol/L Na2SO4.
Figure 4
(a) Removal of NO2−-N at different potentials (100 mL solution with 15 mg/L NO2−-N, 60 min treatment). Plots of −ln(ct/c0) versus time for (b) NO3−-N removal and (c) NO2−-N removal on CuO, CuCoOx, and Co3O4 at −1.1 V. The values in the figure are the corresponding rate constant (k). (d, e) DEMS measurements of the NO3RR on CuCoOx and CuO (three cycles under the potential of −1.1 V). (f) Proposed mechanism of NO3RR on Cu-Co-O catalysts.
Figure 5
(a) The consecutive cycling test at −1.1 V on CuCoOx electrode. (b) The percentage of NO3−-N removal and NH4+-N selectivity on CuCoOx at different pH for 120 min treatment. (c) LSV curves of NO3RR on CuCoOx||Ir-Ru/Ti with a two-electrode configuration. (d) The percentage of NO3−-N removal and NH4+-N generation (left axis) and NH4+-N selectivity (right axis) on CuCoOx||Ir-Ru/Ti at different cell voltage.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
