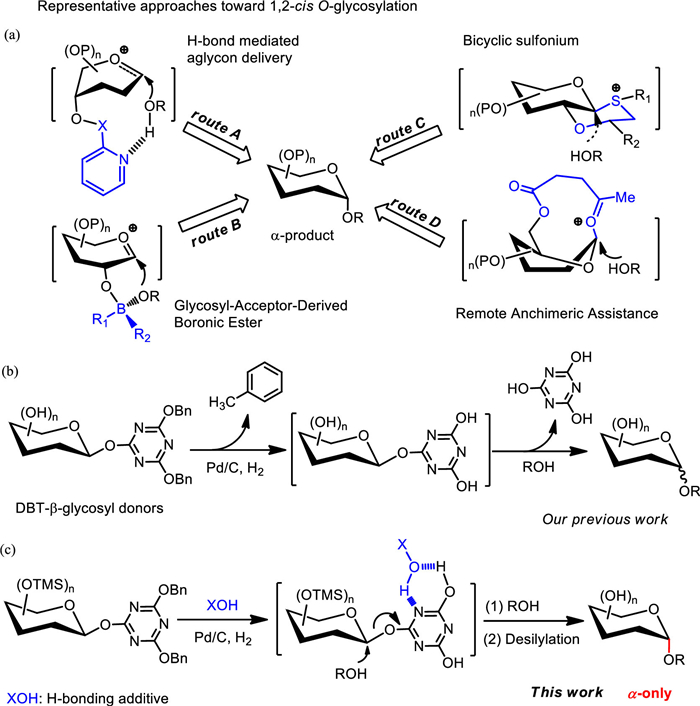
Figure 1.
Introduction and background of current work.
Hydrogen bond-assisted 1, 2-cis O-glycosylation under mild hydrogenolytic conditions
Gefei Li , Yanlong Luo , Juan Mo , Masato Noguchi , Jie Jing , Zhenyang Luo , Shin-ichiro Shoda , Xin-Shan Ye

The diverse 1, 2-cis glycosidic moieties could be commonly found in pharmaceuticals and natural products such as lipids, terpenoids, polyketides [1], in which sugar residues act as a decisive factor in their architectural complexity and bioactivity [2]. Moreover, anomerically alkyl glycosides serving as fundamental building blocks are in demand to construct cell-wall structures [3]. Low abundance and heterogeneity of these glycoconjugates in nature cause the major obstacles in isolation and characterization. Methodologies for the efficient synthesis of homogeneous and structurally well-defined α-glycosyl natural products have thus attracted attention in many research fields to aid in understanding the precise biological roles and structure-activity relationships of these attached sugars. Clearly, uncovering the contributions of carbohydrates to natural medicines and nutraceuticals would greatly facilitate advances of glycosciences.
To meet the needs of stereo-specific synthesis, various strategies were developed to offer a neighboring-group effect or supramolecular interaction, generating the α- and β-specific products [4-6]. Although the 1, 2-cis-α linkage is favored by the anomeric effect, the stereospecific synthesis of 1, 2-cis-α-product is still challenging for chemists. In 1981, the first use of β-glucosyl fluoride as a glycosyl donor was reported for the synthesis of α-glucosides with a promoter generated from stannous chloride (SnCl2) and silver perchlorate (AgClO4) [7]. The poorer leaving group derived from a strong C-F bond leads to the selective formation of α-products. Currently, many advanced and representative protocols including H-bond mediated aglycon delivery (HAD) strategy (Fig. 1a, route A) [8], boronic ester mediated 1, 2-cis-α-glycosylation (Fig. 1a, route B) [9-12], neighboring auxiliary method (Fig. 1a, route C) [13], and remote assistance effect (Fig. 1a, route D) via O-Bz [14], 6-O-Lev protecting group [15, 16], or 1, 6-di-O-N-phenyltrifluoeoacetimidoyl glycosyl donors [17], have been reported to overcome this issue.
In addition, some new methods have also been demonstrated for the efficient synthesis of α-glycosides by using traditional donors, such as phenanthroline-glycosyl bromide system [18], designed triflation of the 2-hydroxyethyl group [19], in-situ adduct transformation with thioglycosides [20], halogenated benzyl protecting groups [21, 22], and gold-catalyzed glucosylation using an O-ethynylphenyl β-D-1-thioglucosyl donor [23]. In a different approach, Niu and co-workers reported a distinct 1, 2-cis-α-glycosylation enabled by halogen-bond-assisted radical activation rather than the conventional ionic process [24]. More recently, the α-selective O-glycosylation reactions towards 2-deoxyglycosides were also successfully established in our group by using electro-chemical strategy [25] and visible-light-promoted protocol [26].
Although the excellent stereoselectivity could be obtained through these advanced strategies, the preparation of special glycosyl donors with designed protecting groups also brings seriously synthetic hurdles, especially when the direct glycosyl coupling reaction of an oligosaccharide moiety is required in the total synthesis of natural products. Thus, simple and stereocontrolled methods for the construction of completely 1, 2-cis-α-glycosidic linkages are still welcome.
In our previous work (Fig. 1b), a protecting group-free 1, 2-cis-α-glycosylation by using DBT-β-glycosyl donors under hydrogenolytic conditions was demonstrated [27]. The preferential α-selectivity might be rationalized by the speculated H-bond-mediated stereospecific manner (SN2 type). However, the α-selectivity was dramatically decreased in the glycosylation with secondary and tertiary alcohols. The weak hydrogen bond interaction between 4, 6-dihydroxy-1, 3, 5-triazin-2-yl (DHT)-β-glycosyl intermediates and alcohols was postulated as the key factor resulting the undesired stereoselectivity. In this context, we want to employ some hydrogen-bonding additives in this hydrogenolytic glycosylation to offer stronger H-bond interaction (Fig. 1c). Obviously improved reaction efficiency and α-selectivity have been noted with assistance of chloral hydrate or hexafluoro-2-propanol (HFIP). We have also disclosed the experimental evidence for the intermolecular hydrogen-bonding effect via proton NMR and DFT calculation.
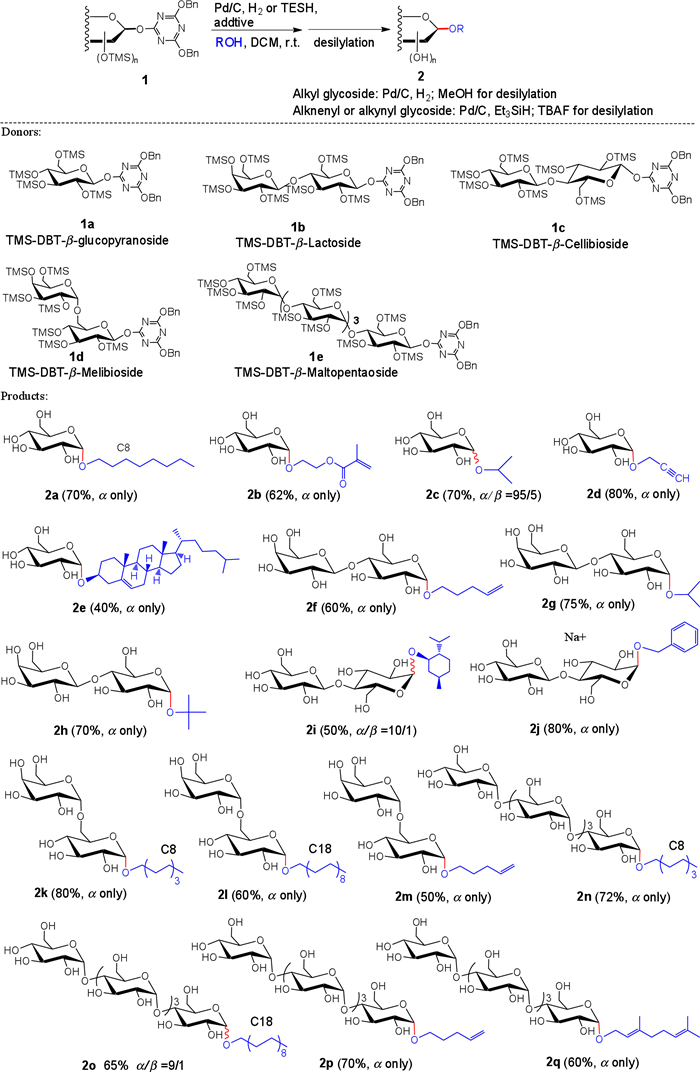
The glycosyl donors were prepared according to our reported work via the direct anomer-activation under aqueous media. Then, the produced DBT-β-glycosyl donors were isolated through simple washing with water and chloroform to remove the native sugars and minor α-type 1, 2-(DBT)2-glycosides (Fig. S2–2 in Supporting information). The protection of trimethylsilyl (TMS) group enables us to synthesize glycosides having hydrophobic moiety, since TMS group acts as solubilizing group in organic solvent like dichloromethane and can be removed under mild conditions like hydrogenolysis. Glycosylation reactions can be carried out under Pd/C (10 wt%)-catalyzed hydrogenolysis conditions, giving rise to the α-glycosides via an SN2-like course. When acceptors containing alkenyl group are used, selective debenzylation and subsequent glycosylation can be achieved by using triethylsilane (TESH) as the mild reductant instead of H2 atmosphere.
Initially, the model glycosylation of TMS-DBT-β-glucopyranoside 1a and 2-propanol was screened with varied solvents (Table 1, entries 1–6). It was found that dichloromethane (DCM) supported the best results for both yield and α-selectivity. As anticipated, a remarkable decrease in yield was observed along with generation of the byproduct B1 when a relatively low amount of 2-propanol was used (Table 1, entry 2). Whereas, the reaction failed in toluene or ethyl acetate due to the inhibited activity of Pd metals which can be strongly coordinated with solvent molecules [28]. Under a solvolysis condition in 2-protanol, the desired α-product was stereospecifically formed in good yield (Table 1, entry 7). Replacing the TMS groups with other protecting groups led to the different results (Table 1, entries 8 and 9). When using triethylsilylated glycosyl donor (TES-DBT-β-glucopyranoside) 1f, a decreased α-selectivity was observed due to the steric hindrance. In case of using O-acetyl donor 1g, the desired glucoside was not obtained, and the orthoester product B2 was generated quantitatively. These results disclose the optimized reaction conditions of TMS-donors in DCM.
According to the postulated mechanism, it is more difficult for the elimination of DHT leaving group in the presence of secondary or tertiary alcohols due to a weaker hydrogen-bonding interaction. To facilitate the departure of DHT group, some additives were added to this reaction to afford a stronger H-bond interaction. In the model reaction, the coupling reaction of 1b was carried out in dry tert-butyl alcohol with the assistance of diverse additives (Table 1, entries 11–18). There is no doubt that low yield was noticed in the absence of additives (Table 1, entry 10). The improved yields were observed with the participation of additives (Table 1, entries 11–18). Among them, the desired α-selectivity was achieved in most cases. However, the reaction with hexachloroacetone gave the product as an α/β-mixture in high yield (Table 1, entry 18). As chloral hydrate (Table 1, entry 15) and hexafluoroisopropanol (HFIP, Table 1, entry 17) have proven to be very effective in this additive-assisted α-glycosylation, the reactions with other substrates are carried out by using these highly promising additives.
With optimal conditions in hand, we next set out to screen the reaction scope. The glycosylation of diverse acceptors with various donors including oligosaccharide-derived donors were performed smoothly under the standard additive-assisted reaction conditions, affording highly α-specific glycosides in moderate to satisfactory yields (Scheme 1). According to this reaction, series of amphiphilic glycolipids, which are widely used as the nonionic biosurfactants in the cosmetic and pharmaceutical industry, could be prepared in good yields. The obtained alkenyl and alkynyl α-glycosides could be employed as reactive precursor for further reactions such as polymerization for the production of glycopolymers. It was noteworthy that challenging secondary and tertiary alcohols were glycosylated in good α-selectivity under the present method (2e, 2g, 2h). The reaction of higher oligosaccharides (maltopentaose: α(1, 4)-Glc5) also successfully occurred in satisfied yields and with high 1, 2-cis-α-selectivity (2n-2q). These results showed attractive applications of this hydrogen-bond-assisted glycosylation to offer an efficient and simple route towards construction of α-glycosidic bonds.
Finally, the reaction mechanism was studied via the NMR experiment and DFT calculation, providing the evidence that hydrogen-bonding interaction between additives and DHT-intermediates is critical for the efficient glycosylation. Normally, it is difficult to monitor the highly reactive intermediate that is very unstable under the ambient conditions. In our experiments, this reaction was able to be inhibited with the addition of amines (e.g., triethylamine), forming the amine-stabilized DHT-glycosyl intermediate, which offers an alternative model to study hydrogen-bonding between additives and intermediates (Fig. 2a). Thus, NMR spectroscopy was performed towards the mixture of amine-stabilized intermediate from 1b and different additives, respectively, to reveal the shifts of the anomeric proton (H-1) signals (Fig. 2b). Since the chemical status of DHT leaving group could influence the electron density of the anomeric center, the chemical shift of anomeric proton might be useful in explaining the observed improvement from additive-assisted glycosylation. Expected shifts (upfield shift of the H-1) could be identified for the mixture of amine-stabilized intermediates and additives in CD2Cl2. Furthermore, the increased upfield shift of the anomeric proton was observed with the decreased pKa values of additives. The magnitude of the chemical shift is thought to be indicative of the H-bond strength. Similarly, another hydrogen-bond-mediated upfield shift of anomeric proton has also been displayed in the thiourea-assisted glycosylation [29].
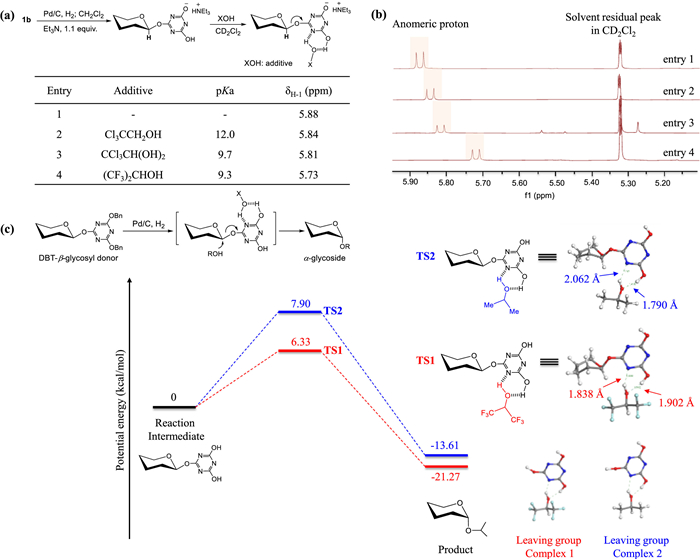
Furthermore, we studied the reaction mechanism via the density functional theory (DFT) code Dmol3 in the framework of the B3LYP exchange-correlation functional (Fig. 2c). The double numerical plus polarization (DNP) was utilized as the numerical basis set during the simulation. The core electrons treatment was implemented by DFT semicore pseudopotential. It should be noted that TS1 derived from DHT-glycosyl intermediate and HFIP has lower free energy barrier (6.33 kcal/mol) than TS2 (7.90 kcal/mol), indicating that the glycosylation could be activated via the hydrogen-bond-induced six-membered ring complex. Stronger hydrogen-bonding is more favorable for a SN2-manner, leading to the production of α-glycosides. These results are consistent with the experimental results.
In summary, a hydrogen bond-assisted α-glycosylation reaction with DBT-β-glycosyl donors was presented. Hydrogenolytic debenzylation gave the glycosyl intermediate with a DHT leaving group which would be further activated by an H-bond interaction with the glycosyl acceptor or additive, offering an α-glycosidic product through SN2 manner. NMR studies confirmed that the chemical shift of anomeric proton in the glycosyl intermediate moved to the upfield due to the H-bond interactions with additives, which indeed facilitated the glycosylation reaction. Density-functional theory calculations were consistent with the experimental results, suggesting a lower free energy barrier in the presence of HFIP additive. It is hoped that this new method will assist glycochemists in the modification of natural products with the α-linked carbohydrate moiety. Further versatility of the present glycosylation is underway.
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the State Key Laboratory of Natural and Biomimetic Drugs (No. K202216), the National Natural Science Foundation of China (Nos. 21907004 and 81821004), the National Key R & D Program of China (No. 2018YFA0507602), the Beijing Outstanding Young Scientist Program (No. BJJWZYJH01201910001001), and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Supplementary material associated with this article can be found, in the online version, at doi:
U. Mrudulakumari Vasudevan, E.Y. Lee, Biotechnol. Adv. 41 (2020) 107550–107572. doi: 10.1016/j.biotechadv.2020.107550
Y. Yang, X. Zhang, B. Yu, Nat. Prod. Rep. 32 (2015) 1331–1355. doi: 10.1039/C5NP00033E
B. Meng, Z. Zhu, D.C. Baker, Org. Biomol. Chem. 12 (2014) 5182–5191. doi: 10.1039/C4OB00626G
S.S. Nigudkar, A.V. Demchenko, Chem. Sci. 6 (2015) 2687–2704. doi: 10.1039/C5SC00280J
X. Qin, X.S. Ye, Chin. J. Chem. 39 (2021) 531–542. doi: 10.1002/cjoc.202000484
W. Yong, X.S. Ye, Acta Chim. Sin. 77 (2019) 581–597. doi: 10.6023/A19040128
T. Mukaiyama, Y. Murai, S. Shoda, Chem. Lett. 10 (1981) 431–432. doi: 10.1246/cl.1981.431
J.P. Yasomanee, A.V. Demchenko, J. Am. Chem. Soc. 134 (2012) 20097–20102. doi: 10.1021/ja307355n
A. Nakagawa, M. Tanaka, S. Hanamura, D. Takahashi, K. Toshima, Angew. Chem. Int. Ed. 54 (2015) 10935–10939. doi: 10.1002/anie.201504182
M. Tanaka, A. Nakagawa, N. Nishi, et al., J. Am. Chem. Soc. 140 (2018) 3644–3651. doi: 10.1021/jacs.7b12108
S. Izumi, Y. Kobayashi, Y. Takemoto, Org. Lett. 21 (2019) 665–670. doi: 10.1021/acs.orglett.8b03823
S. Tomita, M. Tanaka, M. Inoue, et al., J. Org. Chem. 85 (2020) 16254–16262. doi: 10.1021/acs.joc.0c02093
R.A. Mensink, T.J. Boltje, Chem. Eur. J. 23 (2017) 17637–17653. doi: 10.1002/chem.201700908
M. Shadrick, Y. Singh, A.V. Demchenko, J. Org. Chem. 85 (2020) 15936–15944. doi: 10.1021/acs.joc.0c01279
Y. Zhang, Z. Chen, Y. Huang, et al., Angew. Chem. Int. Ed. 59 (2020) 7576–7584. doi: 10.1002/anie.202000992
Y. Zhang, H. He, Z. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 12597–12606. doi: 10.1002/anie.202103826
X. Liu, Y. Song, A. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202201510.
F. Yu, J. Li, P.M. DeMent, et al., Angew. Chem. Int. Ed. 58 (2019) 6957–6961. doi: 10.1002/anie.201901346
S.J. Moons, R.A. Mensink, J.P.J. Bruekers, et al., J. Org. Chem. 84 (2019) 4486–4500. doi: 10.1021/acs.joc.9b00022
J.C. Hu, A.W. Feng, B.Y. Chang, C.H. Lin, K.T. Mong, Org. Biomol. Chem. 15 (2017) 5345–5356. doi: 10.1039/C7OB00839B
D.K. Njeri, C.J. Pertuit, J.R. Ragains, Org. Biomol. Chem. 18 (2020) 2405–2409. doi: 10.1039/D0OB00373E
D.K. Njeri, E.A. Valenzuela, J.R. Ragains, Org. Lett. 23 (2021) 8214–8218. doi: 10.1021/acs.orglett.1c02947
Z. Zheng, L. Zhang, Carbohydr. Res. 471 (2019) 56–63. doi: 10.1016/j.carres.2018.10.010
C. Zhang, H. Zuo, G.Y. Lee, et al., Nat. Chem. 14 (2022) 684–694.
M. Liu, K.M. Liu, D.C. Xiong, et al., Angew. Chem. Int. Ed. 59 (2020) 15204–15208. doi: 10.1002/anie.202006115
K.M. Liu, P.Y. Wang, Z.Y. Guo, et al., Angew. Chem. Int. Ed. 61 (2022) e202114726.
M. Ishihara, Y. Takagi, G. Li, M. Noguchi, S. Shoda, Chem. Lett. 42 (2013) 1235–1237. doi: 10.1246/cl.130646
T. Ikawa, K. Hattori, H. Sajiki, K. Hirota, Tetrahedron 60 (2004) 6901–6911. doi: 10.1016/j.tet.2004.05.098
Y. Geng, A. Kumar, H.M. Faidallah, et al., Angew. Chem. Int. Ed. 52 (2013) 10089–10092. doi: 10.1002/anie.201302158

Scheme 1 Substrate scope of the present glycosylation. Reaction conditions: ROH (10 equiv.) for the primary alcohols; ROH (20 equiv.) for the secondary and tertiary alcohols; TESH (5–10 equiv.) was employed as the reductant in case of using unsaturated acceptors; 24 h reaction for the Pd-H2 conditions; 48 h reaction for the Pd-TESH conditions. Chloral hydrate was employed as additive in the synthesis of 2c, 2e and 2i, HFIP was used for the other reactions.

Figure 2 Reaction mechanism studies. (a) Relations between the chemical shifts of the anomeric protons on amine-stabilized DHT-glycosyl intermediates and varied additives; (b) Stacked NMR spectra for the mixture of amine-stabilized DHT-glycosyl intermediates and different additives; (c) DFT calculation results about this reaction: reaction intermediate is generated via hydrogenolytic debenzylation. Then, HFIP and 2-prpanol were added to form the corresponding transition structures TS1 and TS2, respectively. The simulated details and original energy for DFT calculation were listed in Supporting information.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载: