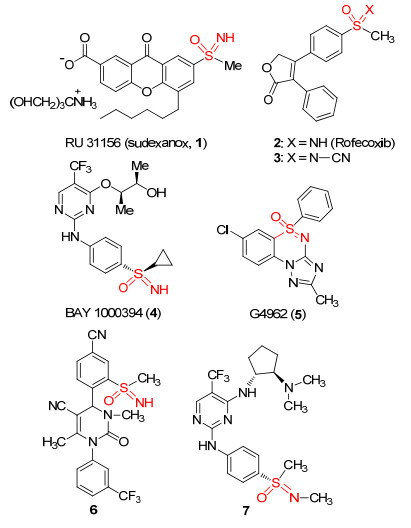
图 1.
一些含有亚氨基砜骨架的药物分子
Figure 1.
Some pharmaceutical molecules containing sulfoximine core structure

亚氨基砜是一类重要的含硫氧双键和硫氮双键的化合物, 其合成最早由Whitehead和Bentley等[1]于20世纪50年代报道, 亚氨基砜也是一种重要的化工原料和有机合成药物中间体, 同时也可以作为一种良好的含氮导向基团用于碳氢键的直接官能团化反应, 或者作为手性辅助剂或配体应用在不对称合成中[2].
含有亚氨基砜结构的化合物具有多种生物活性, 如杀虫活性、杀菌活性等, 因此被广泛应用于农药和医药等领域以及用作药效基团应用于在药物化学研究(图 1).例如, 引入亚氨基砜基团的口服药物RU31156 (1), 能够通过抑制肥大细胞组织胺上过敏物质的释放而用作临床治疗过敏性或运动诱发性哮喘[3].带有砜结构非甾体抗炎药Rofecoxib (2)能选择性抑制COX-2, 但是长期服用对心血管有副作用.而其N-氰基取代的类似化合物3则能大大降低副作用[4]. BAY1000394 (4)是一种强效的抗增殖活性的纳米级pan-CDK抑制剂[5]. G 4962 (5)是一种高效的抗焦虑、抗惊厥的苯二氮卓类受体激动剂[6].化合物6作为一种新型的人中性粒细胞弹性蛋白酶抑制剂, 可以治疗或预防肺部及心脑血管疾病[7].化合物7是一种酪氨酸激酶抑制剂[8], 可以用于治疗骨质疏松.此外, 由于亚氨基砜与醇、酸、脒、砜、酰胺等化合物互为电子等排体, 且其在生物医疗方面有潜在作用, 因此, 研究高效合成含有亚氨基砜骨架的化合物具有重要的理论意义与应用前景.
由于亚氨基砜系列衍生物在生物医药、农药和有机合成等领域中都有广阔的应用前景, 引起了有机合成化学家和药物合成化学家的广泛关注.近年来, 合成化学家对亚氨基砜的合成方法及其在有机合成中的应用展开了大量研究, 取得了丰硕的研究成果.前期的文献综述了近期的一些典型的含有亚氨基砜结构的新型药物[9].然而, 对于如何制备或获取这些小分子化合物的综述性文献却鲜有报道.尤其是近年来, 各种合成亚氨基砜类化合物的新方法层出不穷, 亚氨基砜合成方法的发展有力地推动相关反应活性的研究, 拓展了亚氨基砜在有机合成中的应用.基于此, 本文主要综述了近五年来亚氨基砜衍生物的合成方法及其在有机合成中的应用研究进展.
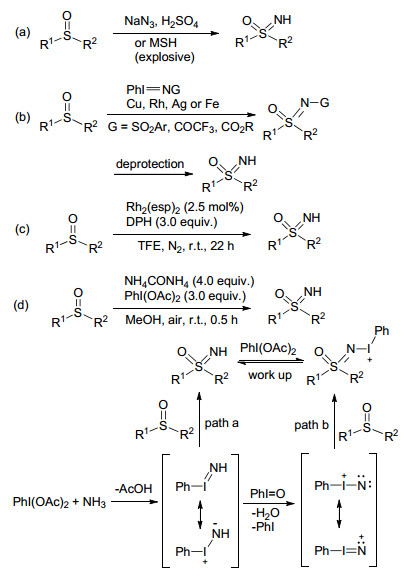
对亚砜进行胺化是制备NH-亚氨基砜的常用方法, 但传统方法需要用到叠氮化钠、浓硫酸、O-三甲基苯磺酰羟胺(MSH)等有毒或易爆的危险性试剂[10, 11](Scheme 1, a).为了克服这一缺点, 近年来也发展了一些过渡金属催化的亚砜胺化反应, 但大多数是生成N-保护的亚氨基砜, 得到NH-亚氨基砜则需要额外的脱保护过程(Scheme 1, b)[12], 因此发展一种温和简便的方法来制备NH-亚氨基砜已成为科学家们目前研究的热点. 2014年, Ge等[13]报道了首例铑催化的亚砜为底物的直接合成NH-亚氨基砜的新方法(Scheme 1, c), 该方法以O-(2, 4-二硝基苯基)羟胺(DPH)作为胺化剂, 室温下就能完成转化, 而且具有很好的官能团容忍性. 2016年, Bull课题组[14]实现了在无需任何过渡金属催化下的亚砜胺化反应(Scheme 1, d), 该反应以氨基甲酸铵作为氮源, 与醋酸碘苯按比例混合, 通过产生高亲电性中间体PhINH或PhIN+, 快速地与亚砜发生亲电反应得到NH-亚氨基砜.
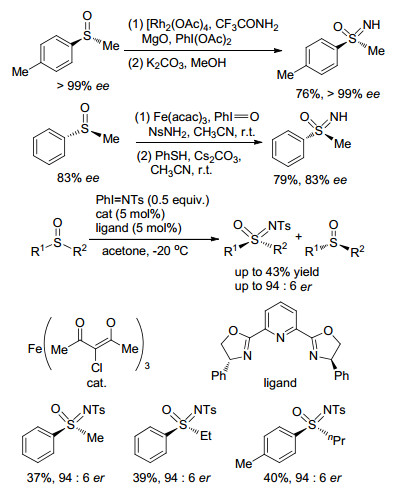
从高光学纯度的亚砜底物出发, 通过过渡金属催化胺化, 是早期合成手性亚氨基砜的常用方法之一.例如, 2004年, Bolm课题组[12d]以Rh2(OAc)4作催化剂, 三氟乙酰胺与醋酸碘苯按比例混合作胺化剂, 实现了手性亚氨基砜的合成(Scheme 2); 2006年, Olga等[12f]报道了Fe(acac)3催化、亚碘酰苯氧化下的磺酰胺对手性亚砜的胺化反应, 在较短时间内可以得到构型保持的亚氨基砜产物(Scheme 2). 2014年, Bolm课题组[15]发现了一种合成手性亚氨基砜的新方法, 该方法以外消旋的亚砜为底物, PhI=NTs为氮烯前体, 在手性铁催化剂的作用下, 通过动力学拆分能得到手性亚氨基砜产物(Scheme 2).他们发现增强Fe(Ⅲ)中心路易斯酸酸性可以提高自身的催化活性和反应的对应选择性, 反应的底物适应性较好, 各种外消旋的亚砜都能在反应中给出良好的收率和对映选择性.
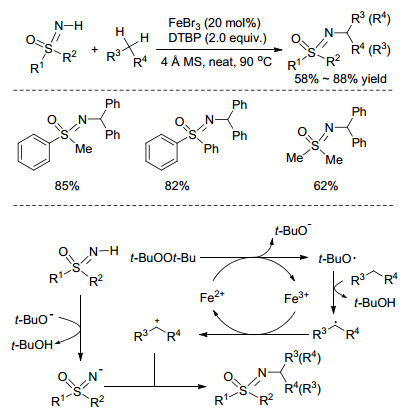
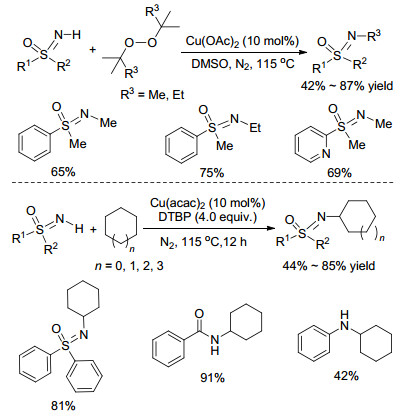
2014年, Bolm课题组[16]以FeBr3作催化剂、二叔丁基过氧化物(DTBP)作氧化剂, 在无溶剂条件下高效构建了C(sp3)—N键, 实现了NH-亚氨基砜与二芳基甲烷C(sp3)—H键的脱氢氧化偶联反应.作者通过底物拓展实验发现, 不对称的二芳基甲烷参与反应将得到比例为1:1的对映异构体, 由此认为该反应可能经历了自由基过程: Fe2+诱导DTBP分解生成叔丁氧基自由基, 该自由基夺取苄基C(sp3)—H上的氢, 进而产生二苯甲基自由基, 再经过单电子转移过程生成苄基正离子, 最后与去质子化的亚胺负离子结合生成最终产物(Scheme 3).
Yu课题组[17]报道了以Cu(OAc)2作为催化剂, DTBP直接作为甲基源的亚氨基砜的N-甲基化反应(Scheme 4), 值得注意的是, 使用二(二甲基丙基)过氧化物作为烷基源时可以发生N-乙基化反应, 目标产物的收率为60%~81%. 2015年, Cheng课题组[18]利用环己烷/环庚烷等简单的惰性烷烃作为底物, 实现了与亚氨基砜的脱氢偶联反应(Scheme 4).该反应底物适应性良好, 对芳基酰胺, 芳胺类等底物同样适用.该反应同样可能经历了自由基过程, 环己烷首先被氧化成环己基自由基, 然后与亚氨基砜负离子结合而形成最终产物.
Bolm课题组[19]以三氟甲基三甲基硅烷(TMSCF3)为偶联试剂, 通过Ag+催化N—H/Si—CF3交叉偶联合成N-三氟甲基亚氨基砜化合物.研究表明Ag2CO3作为催化剂, 1, 10-菲罗啉作为配体的催化体系能有效促进反应的进行, 而1, 4-二氮杂二环[2.2.2]辛烷(DABCO)或2, 9-二甲基-1, 10-菲罗啉等其它配体的加入均不能活化反应体系(Scheme 5).
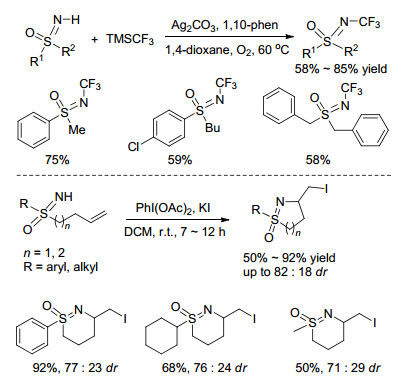
2016年, Bolm课题组[20]报道了无过渡金属参与的烯基的双官能化反应, 在醋酸碘苯[PhI(OAc)2]的氧化作用下, 一系列的S-烯基-NH-亚氨基砜在碘化钾(KI)的促进下发生反应, 可以高区域及高非对映选择性地得到5-exo或6-exo型碘环化产物(Scheme 5).可能是位阻的影响, 若底物碳-碳双键上连有两个以上的取代基, 反应活性很低甚至不反应.
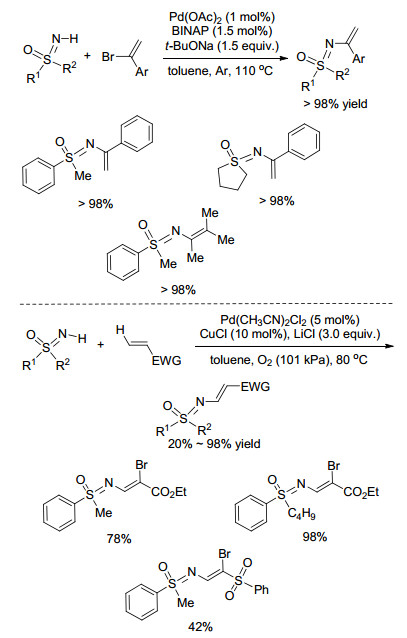
含有烯基的化合物化学性质较为活泼.由于烯基具有高的转化能力(加成、取代、氧化反应等), 是有机合成中最基本的合成砌块, 因此在生物活性分子中引入烯基一直是有机合成工作者的研究热点, 在亚氨基砜的N原子上引入烯基基团有很好的应用前景. 2004年, Bolm课题组[21]发展了一种钯催化的NH-亚氨基砜与α-溴代苯乙烯的取代反应, 反应在1 mol%的Pd(OAc)2催化下便可高效地实现NH-亚氨基砜N-烯基化(Scheme 6).值得一提的是, 该体系适用于具有空间位阻效应的2-溴-3-甲基-丁-2-烯, 然而对β-苯乙烯并不适用, 这可能是由于与β-苯乙烯反应所得的产物不稳定易分解的原因.该课题组在之前工作和理论基础上, 使用一系列烯丙基酯作为底物, 在Pd/Cu共催化和LiBr存在条件下, 与NH-亚氨基砜发生烯烃胺化溴化反应[22], 以良好至优秀的收率得到Z型双官能化产物(Scheme 6).作者对丙烯酸酯衍生物底物进行了拓展, 发现丙烯酸酯的酯基上带有吸电子集团(氰基、酯基等)时, 目标产物产率会降低, 推测其原因可能是吸电子基团参与了与金属的配位, 降低了催化剂的催化能力.
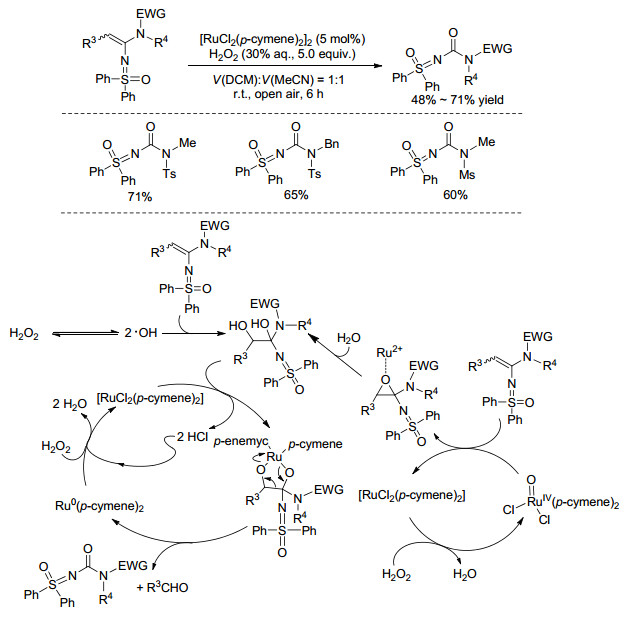
最近, 利用基于NH-亚氨基砜的弱亲核性氮元素与炔烃的加成反应也有报道. Chen课题组[23]成功发展了PPh3AuCl和AgOTf共催化的亚氨基砜的N—H键对炔胺的分子间加成(Eq. 1).该体系条件温和, 底物适用范围很广, 不仅适用于带各种给电子基或吸电子基的芳基炔胺, 还适用于烷基炔胺和噻吩炔胺, 这些底物都能高效地构建N-烯基亚氨基砜类产物.有意思的是, 该反应产物可以进一步发生烯键的选择性氧化切断反应, 在[RuCl2(p-cymene)2]2/H2O2的催化氧化体系下, 发生碳碳双键的断裂, 得到非对称的亚氨基砜脲类衍生物.在这一过程中四甲基哌啶氮氧化物(TEMPO)或2, 6-二叔丁基苯酚(BHT)的加入会显著抑制反应的进行, 因此推测此氧化反应可能经历了自由基过程(Scheme 7).
|
|
(1) |
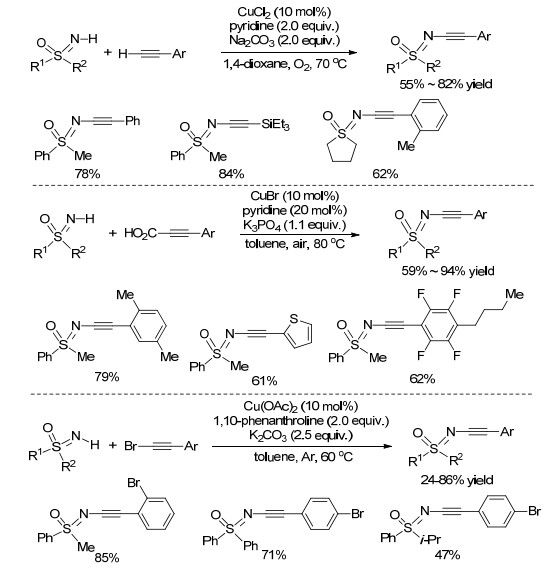
炔烃是有机合成中一类重要的结构单元, 广泛存在于药物、天然产物及材料中. N-炔基亚氨基砜是合成亚氨基砜手性配体的重要中间体, 炔基的存在也为构建新型亚氨基砜类化合物提供了更多的可能. 2013年, Bolm课题组[24]报道了首例Cu催化NH-亚氨基砜与末端炔烃交叉脱氢偶联构建N—C(sp3)键的新方法(Scheme 8).该反应只对末端炔烃有效, 使用溴乙炔基苯只能得到二炔自身偶联产物, 芳环上连有给电子基团、吸电子基团及卤素原子的末端炔烃都能与NH-亚氨基砜反应, 得到中等收率的产物.遗憾的是, 脂肪族的末端炔烃在该体系中不反应.随后, 该课题组[25]用类似的条件, 将吡啶的用量降至0.2 equiv., 实现了NH-亚氨基砜和芳基炔丙酸的脱羧偶联反应(Scheme 8).不足之处是该方法底物适应性较差, 丙炔酸芳环上连有给电子基团时, 产率会大大降低. 2014年, Bolm课题组[26]在之前两项的工作基础上, 报道了Cu催化NH-亚氨基砜和溴乙炔基苯的直接交叉偶联构建N-炔基亚氨基砜化合物, 为N-炔基亚氨基砜的合成提供了一条新途径(Scheme 8), 反应无需外部氧化剂(空气或氧气), 仅在惰性气体氛围下即可高收率地得到产物.
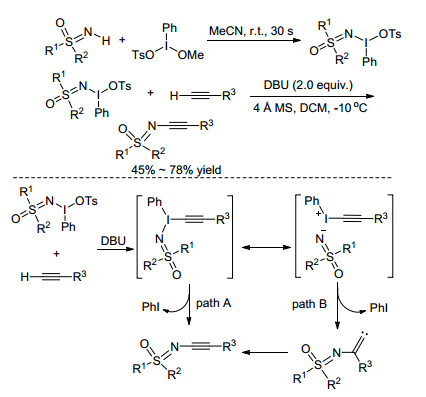
利用含氮高碘试剂构建分子间的C—N键是有机合成中的重要方法. 2016年, Bolm课题组[27]发展了一种无过渡金属催化的合成N-炔基亚氨基砜的新方法.该反应分两步进行:首先在乙腈溶液中, NH-亚氨基砜与甲氧基(甲苯磺酰氧基)碘代苯(MTIB)发生亲核取代反应, 几乎定量地生成新型亚氨基砜高碘试剂; 然后在二氯甲烷溶液中在1.8-二氮杂二环[5.4.0]十一烷-7-烯(DBU)的促进下, 该试剂与苯乙炔在低温条件下反应, 高收率地得到N-炔基亚氨基砜类化合物(Scheme 9).对反应机理的研究表明在碱性条件下, 末端炔烃质子化后与高碘试剂反应生成中间体, 然后此中间体可能经历两种途径, 一种是直接脱去碘苯得到最终产物; 另一种是其互变异构体先脱去碘苯形成亚甲基卡宾, 从而生成最终产物(Scheme 9).
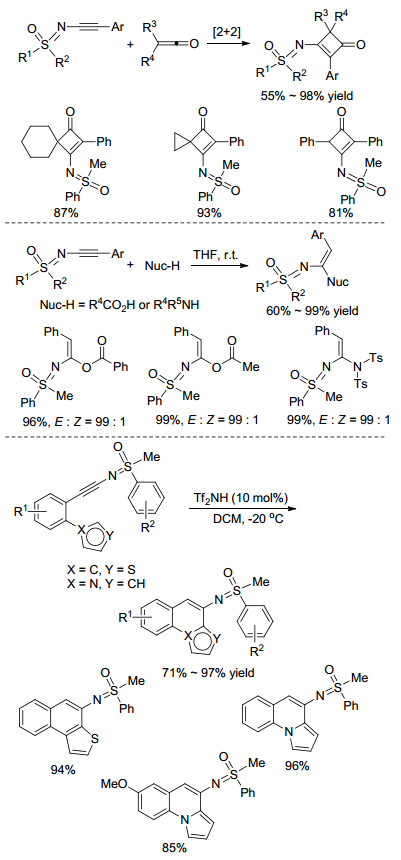
N-炔基亚氨基砜在有机合成上具有十分重要的应用. 2013年, Bolm课题组[28]报道了无金属参与的N-炔基亚氨基砜与烯酮类化合物的[2+2]环加成反应, 得到含亚氨基砜基的环丁酮类化合物(Scheme 10). 2015年, 该课题组[29]报道了苯甲酸类化合物中的酯氢键对炔基的分子间加成, 该反应立体选择性非常好, 当使用四氢呋喃(THF)作溶剂时, 反应几乎只得到E型产物.值得一提的是, 在二氯甲烷(DCM)溶液中时, 使用磺酰胺作为亲核试剂也能同等效率地得到氢化胺化产物(Scheme 10), 这两种氢化产物又可以进一步被氧化成相应的1, 2-二酮类化合物.最近, 利用布朗斯特酸催化的N-炔基亚氨基砜的分子内环化反应也有报道[30], 在双三氟甲磺酰亚胺(Tf2NH)的催化下, 能以6-endo-dig关环的方式高收率获得含亚氨基砜基的萘并噻吩或吡咯并喹啉类化合物(Scheme 10).
2000年, Hildebrand课题组[31]报道了钯催化的NH-亚氨基砜与卤代芳基的交叉偶联反应.碱性条件下, Pd(OAc)2搭配双齿类配体, 可促进NH-亚氨基砜与溴代芳基的反应, 得到N-芳基亚氨基砜(Eq. 2).作者发现添加LiBr, 带吸电子基的芳基碘代物也能以中等收率得到芳基化产物.当选用1, 2-二溴苯或1, 3-二溴苯作底物时, 只能得到单偶联产物, 据此推测反应历程上可能是亚氨基砜基团使第二个溴原子进行氧化加成过程而失活.
|
|
(2) |
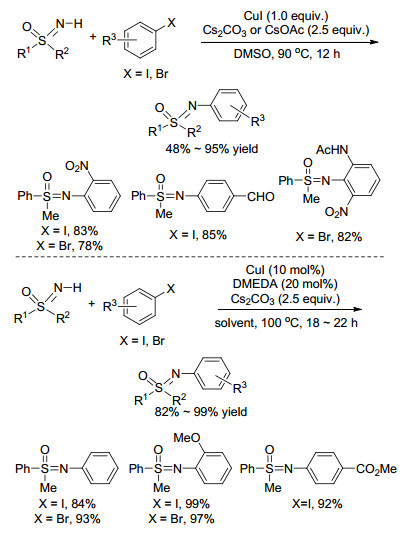
在这之后, 基于NH-亚氨基砜与卤代芳烃的芳基化偶联反应相继被报道.例如, 2004年, Cho等[32]用首次用相对价格低廉的铜作催化剂实现了这一过程(Scheme 11).与钯催化相比, 铜催化的方法能大大缩短反应时间, 并且底物兼容性更好, 各种溴代、碘代芳烃都能较好地适用于该偶联过程.不过令人遗憾的是, 该反应需要使用等物质的量的铜催化剂.基于此, 该组通过进一步研究发现, 加入配体后, 催化量的铜就能促进NH-亚氨基砜与卤代芳烃的偶联[33], 高效构建N-芳基化亚氨基砜类化合物(Scheme 11).需要指出的是, 碘代芳烃作为芳基源时, 可以在甲苯溶剂中一步完成反应; 溴代芳烃作为芳基源时, 需在1, 4-二氧六环溶剂中分两步进行, 反应第一步发生Finkelstein反应, 需要额外添加4.0 equiv. NaI促进反应, 此时芳基上的溴原子被碘原子取代, 第二步加入偶联体NH-亚氨基砜完成反应.
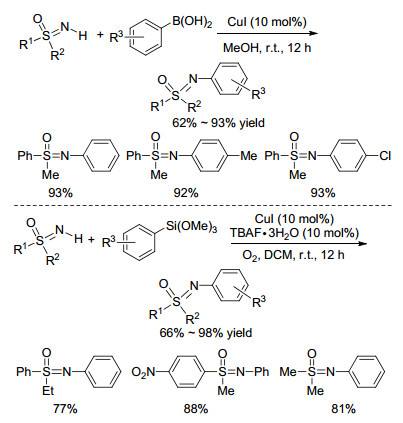
2005年, Bolm课题组[34]发现, 只需要在铜的催化下, 芳基硼酸就可以在温和条件下与NH-亚氨基砜发生偶联, 反应不受电子效应和位阻效应的影响, 能高收率地得到一系列芳基取代的亚氨基砜(Scheme 12). 2014年, Lee课题组[35]在CuI/四丁基氟化铵(TBAF)的催化体系下, 实现了芳基硅烷与NH-亚氨基砜的交叉偶联(Scheme 12), 该方法反应条件温和, 使用氧气作为绿色氧化剂, 可以扩大到克级别, 具有良好的实用性. TBAF在该体系中至关重要, 但是其作用机理尚不明确.
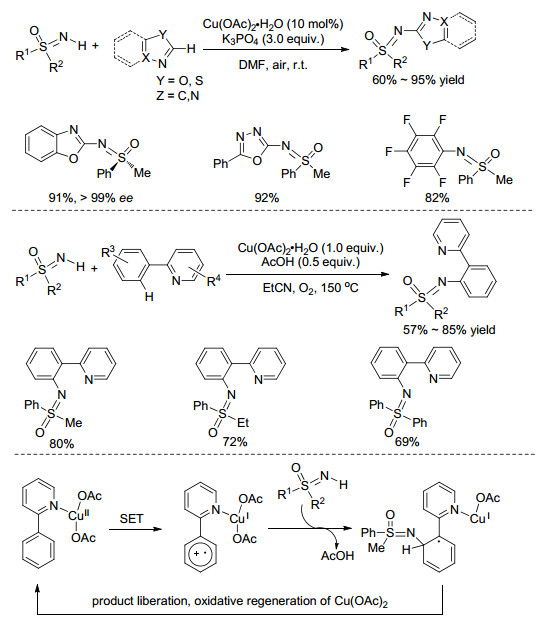
2010年, Miura课题组[36]报道了Cu(OAc)2催化的NH-亚氨基砜和杂环芳烃的脱氢偶联反应(Scheme 13).反应条件温和, 底物范围广, 噁二唑、苯并噁唑、苯并噻唑以及多氟吡啶都能与NH-亚氨基砜反应.值得一提的是, 手性S-甲基-S-苯基亚氨基砜(>99% ee)参与反应后, 可以91%的产率得到手型保持产物(>99% ee). 2014年, Wang等[37]使用2-芳基吡啶作为偶联试剂实现了N-芳基亚氨基砜的合成(Scheme 13), 反应需要氧气的参与, 所得产物作为一种新型N, N-双齿配体有十分潜在的研究意义.作者根据ESI-MS分析出的碎片离子, 提出了可能的反应机理, 首先2-芳基吡啶与金属配位, 然后经历单电子转移过程生成自由基正离子中间体, 紧接着该中间体与NH-亚氨基砜作用形成自由基, 最后被氧化得到最终产物(Scheme 13).
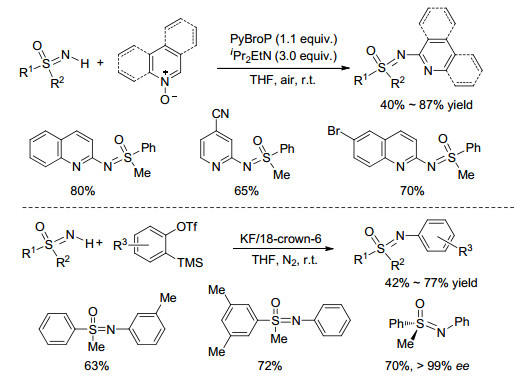
2016年, Singh课题组[38]利用亚氨基砜氮原子的亲核性, 实现了与系列缺电子体系异喹啉氮氧化合物、喹啉氮氧化合物、吡啶氮氧化合物等的偶联反应(Scheme 14, a).该反应无需添加任何过渡金属催化剂, 需加入PyBroP分子来活化氮氧化合物, 在室温下便可高效无污染地实现N-芳基化过程.文中指出, 当选用手性亚氨基砜参与反应时, 可以高收率地得到手性产物, 其ee值高达99%.该课题组[39]报道了在KF/18-冠-6的催化体系下, 2-(三甲基甲硅烷基)苯基三氟甲磺酸酯与NH-亚氨基砜的N-芳基化反应(Scheme 14).芳炔前体在KF作用下产生的高活性中间体芳炔后很容易与亲核试剂发生反应, 其优点在于常温下4 h就能快速完成反应.
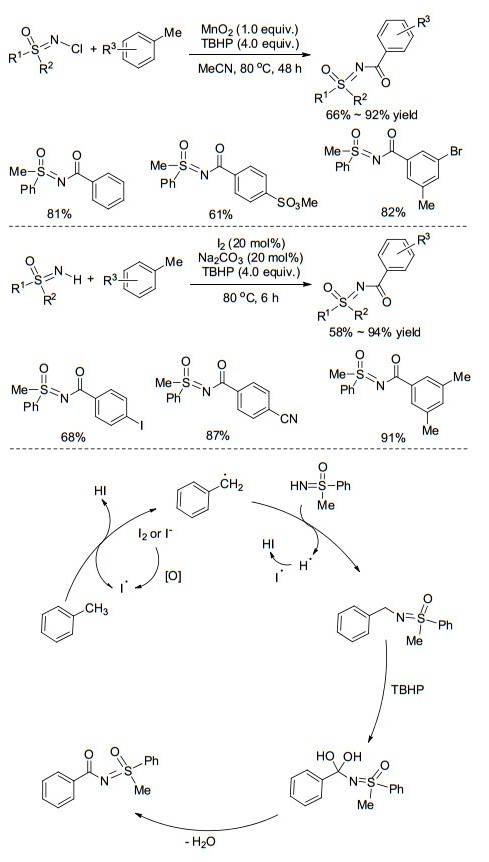
近年来, 在氧化剂存在下, 芳基甲烷可以作为羰基前体参与到各种羰基化偶联反应中. 2014年, Bolm课题组[40]报道了甲基芳烃在氧化条件下与N-Cl亚氨基砜的交叉偶联反应(Scheme 15, a).该反应分两步进行, 首先使用N-氯代丁二酰亚胺(NCS)对NH-亚氨基砜进行氯化, 室温反应2 h后再加入二氧化锰、叔丁基过氧化氢等氧化剂和甲苯, 高温长时间反应可高收率地得到N-酰基化产物.该一锅法反应同时实现了两步反应, 且无需分离氯化的亚氨基砜中间体, 在一定程度上提高了反应效率和反应的步骤经济性. 2015年, An课题组[41]直接以NH-亚氨基砜为底物, 在I2的催化下成功地实现了与芳基甲烷的偶联反应(Scheme 15), 高收率地得到了N-酰基化产物, 拓展了此类反应的底物适用范围, 但对杂环芳烃仍不适用.研究发现, 体系中四甲基哌啶氮氧化物(TEMPO)的加入会抑制反应的进行, 并捕捉到苄基自由基中间体, 因此作者认为反应有可能是甲基芳烃在氧化条件下产生苄基自由基中间体进行的(Scheme 15).
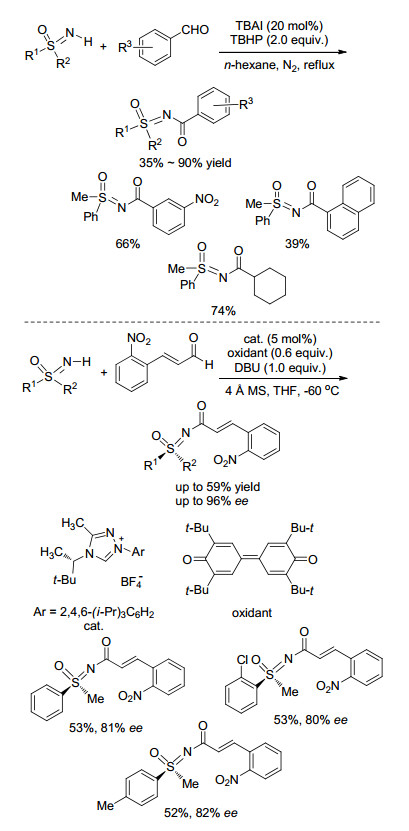
2015年, Deng课题组[42]报道了在季铵盐的催化下, 苯甲醛类化合物在过氧叔丁醇(TBHP)的氧化下可与NH-亚氨基砜发生偶联反应(Scheme 16), 以中等至良好的收率得到一系列N-苯甲酰基亚氨基砜产物. 2016年, Bolm课题组[43]用手性三氮唑盐作为氮杂环卡宾催化剂前体, 在等物质的量的DBU作用下, 消旋的亚氨基砜类化合物可与肉桂醛类底物发生偶联反应(Scheme 16), 高对映选择性地实现了亚氨基砜N-酰基化, 未反应的亚氨基砜也会有旋光性, 其ee值和反ee值分别高达99%和97%.
最近, 我们课题组[44]以苯甲酰基叠氮作为酰基化试剂, 成功实现了与NH-亚氨基砜化合物的偶联(Eq. 3), 反应无需任何催化剂、添加剂, 苯甲酰基叠氮类化合物在加热条件下很容易发生Curtius重排生成高活性的异氰酸酯物种, 再接受NH-亚氨基砜的亲核进攻, 可以快速高效地得到N-酰胺基亚氨基砜产物.产物结构中同时含有亚氨基砜基团和脲骨架, 具有重要的生物医学研究意义.
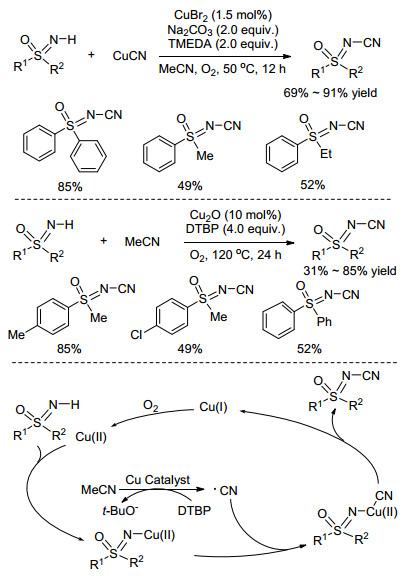
N-氰基(CN)键广泛存在于生物活性分子、天然产物和药物分子中, 目前, N-氰基亚氨基砜在合成方法上的创新也逐渐受到科学家们的关注. 2014年, Cheng课题组[45]以CuBr2作催化剂、发展了一种氰化亚铜(CuCN)与NH-亚氨基砜化合物的偶联方法(Scheme 17), 反应选用乙腈作溶剂, 对照实验证明乙腈不会作为氰基源参与反应.不久之后, Shao课题组[46]报道了铜催化的NH-亚氨基砜与乙腈的偶联反应, 遗憾的是, 该体系需要在氧化条件下高温长时间反应, 限制了其在工业上的应用(Scheme 17).该反应是一个自由基历程, 一方面, Cu2O和TBHP诱导乙腈发生C—CN键断裂产生氰基自由基, 另一方面, Cu(Ⅰ)被氧气氧化成Cu(Ⅱ), 再与亚氨基砜分子配位, 氰基自由基与其发生氧化加成, 最后经还原消除得到产物(Scheme 17).
|
|
(3) |
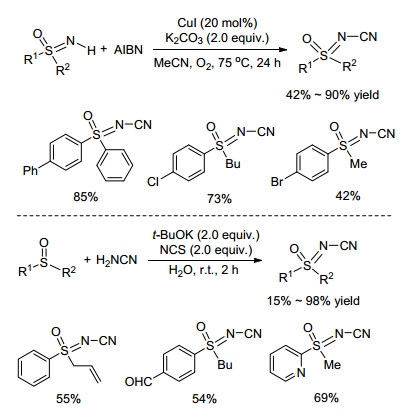
2015年, Teng课题组[47]对铜催化的亚氨基砜N-氰基化反应进行了改进, 采用绿色环保的偶氮二异丁腈(AIBN)作为氰基源, 很大程度上改善了氰化物与NH-亚氨基砜的偶联反应(Scheme 18). 2017年, Bolm课题组[48]从亚砜化合物出发, 在NCS作氧化剂, 腈胺作氰基源条件, 通过一锅法快速高效地一步构建N-氰基亚氨基砜化合物(Scheme 18).值得一提的是, 该反应无需过渡金属催化剂, 且以水作溶剂, 为N-氰基亚氨基砜的合成提供了绿色无污染的新方法.
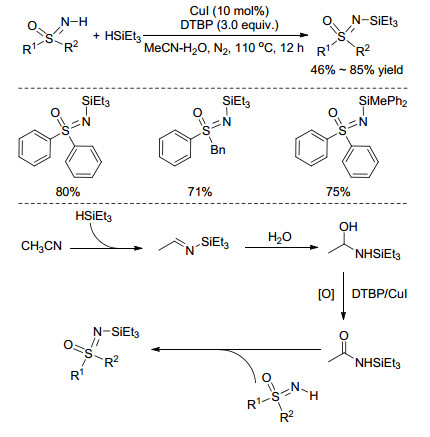
2015年, Cheng课题组[49]报道了铜催化的硅试剂与NH-亚氨基砜的偶联反应(Scheme 19), 可以高收率、高对映选择性地合成一系列N-硅基亚氨基砜化合物.通过气相质谱(GC-MS)监测反应, 发现反应中存在N-三甲基硅基乙酰胺物种.通过同位素标记实验, 他们证实了乙腈作为溶剂参与了反应, N-三甲基硅基乙酰胺中的羰基氧来源于体系中的水(Scheme 19).
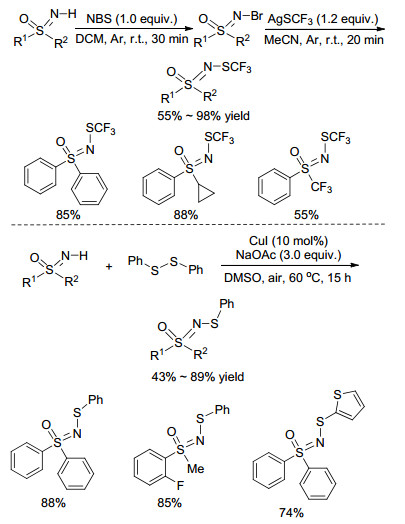
2015年, Bolm课题组[50]先对NH-亚氨基砜进行溴化, 然后以三氟甲硫基银为三氟甲硫基源, 在温和条件下快速实现了亚氨基砜N-硫三氟甲基化反应(Scheme 20). 2016年, Cheng等[51]发现二芳基二硫化物在铜催化下能与NH-亚氨基砜发生偶联反应, 合成一系列硫代亚氨基砜衍生物, 作者推测其反应机理为二芳基二硫化物与Cu(Ⅰ)配位形成硫离子中间体, 然后接受亚氨基砜分子的亲核进攻完成转化(Scheme 20).
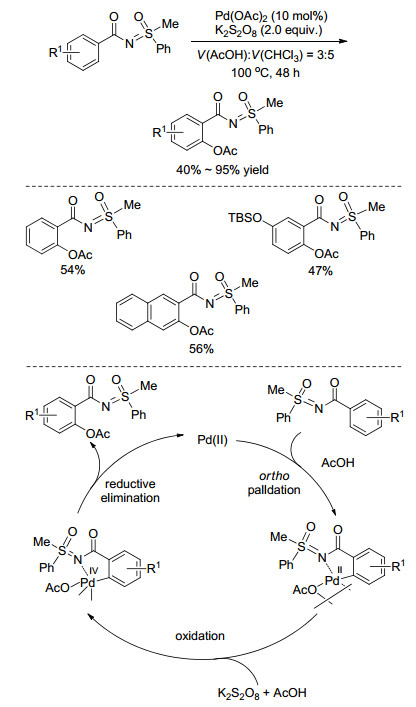
过渡金属催化功能基导向的C—H活化是近年来有机化学领域较为热门的研究方向.在众多导向基团中, 亚氨基砜基作为一种新颖有潜力的定位基引起了有机科学家越来越广泛的关注.利用Pd催化亚氨基砜基导向实现远程C(sp2)—H或C(sp3)—H活化目前为止取得了一些进展. 2012年, Sahoo课题组[52]以N-苯甲酰基-S-甲基-S-苯基亚氨基砜为底物, 在Pd(OAc)2/K2S2O8的催化条件下, 可以实现苯甲酰基芳环上邻位C—H乙酰氧基化(Scheme 21), 产率中等至良好.研究发现溶剂对反应影响很大, 若直接将乙酸作为溶剂, 主要得到邻位单乙酰氧基化后的脱乙酰基产物; 而在乙酸和氯化溶剂组成的混合溶剂中, 则以乙酰氧基化产物(单取代和双取代)为主, 其中在乙酸和和氯仿的体积比为2:3的混合溶剂中的反应效果最好.值得一提的是, 反应对于手性亚氨基砜的底物, 可以获得>99%的极高的对映选择性.作者推测反应经历了Pd(Ⅱ)/Pd(Ⅳ)的转变过程, 首先, Pd(OAc)2与亚氨基砜形成五元环环钯中间体, 然后在氧化剂K2S2O8中与乙酸氧化加成形成Pd(Ⅳ)中间体, 最后经还原消除得到乙酰氧基化产物(Scheme 21).
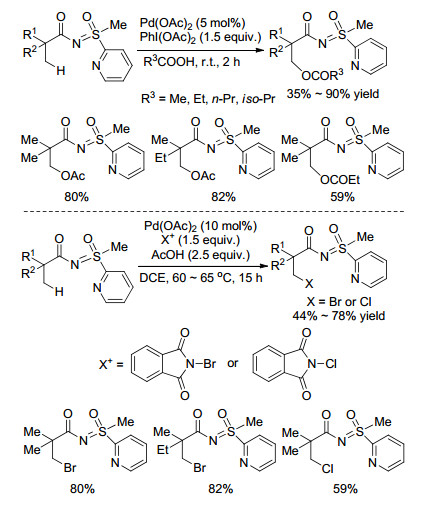
该课题组[53]进一步研究发现, 若将导向基团结构换成甲基吡啶基亚氨基砜, 吡啶基和亚胺基的氮原子可以同时与金属钯发生配位, 能远程诱导N-酰基β位C(sp3)—H活化, 当金属钯和氧化剂同时存在, 溶剂依次为乙酸、丙酸、正丁酸时, 将分别得到β碳乙酰氧基、丙酰氧基、正丁酰氧基化产物(Scheme 22). 2014年, 该课题组[54]选用相同的底物, 以N-卤代(溴、氯)邻苯二甲酰亚胺为卤化试剂, 在Pd(OAc)2催化下实现了β位C(sp3)—H卤化反应(Scheme 22).
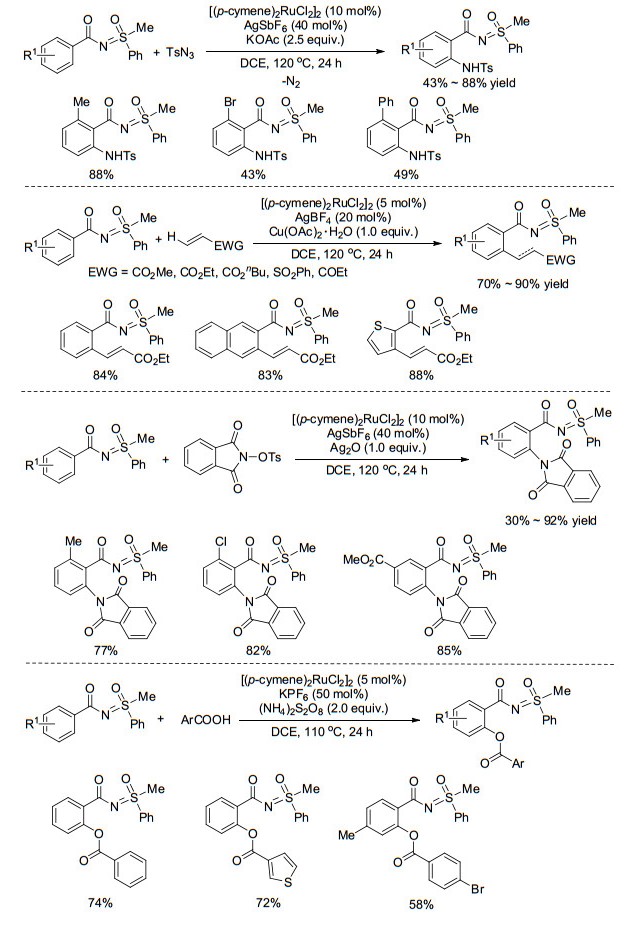
相对于Pd催化而言, 以N-苯甲酰基-S-甲基-S-苯基亚氨基砜为底物, 利用相对廉价的Ru催化剂对苯甲酰基芳环上的邻位进行C—H活化的研究较为广泛. 2013年, Sahoo课题组[55]首先以TsN3为氮源, [(p-cymene)2-RuCl2]2为催化剂, 成功实现了上述化合物苯甲酰基芳环上的邻位胺化反应(Scheme 23).该反应有很好的化学选择性和立体选择性, 唯一的副产物为N2, 符合绿色化学的要求.随后, 陆续有课题组报道了类似底物苯甲酰基芳环上邻位烯基化反应[56](Scheme 23)、邻苯二甲酰亚胺化反应[57](Scheme 23)及酯基化反应[58](Scheme 23), 基本的反应过程为亚氨基砜氮原子与金属Ru发生配位, 接着发生芳基邻位C—H活化形成环钌中间体, 紧接着与各种偶联试剂发生氧化加成, 最后经还原消除步骤得到相应产物.
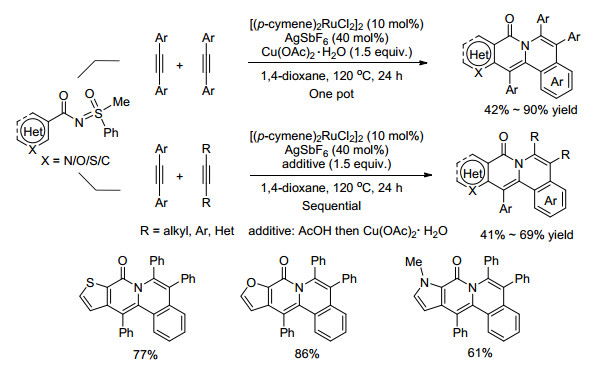
与上述反应不同的是, 在[(p-cymene)2RuCl2]2催化作用下, 当底物分子发生C—H活化的芳烃为杂环芳烃(呋喃、吡咯、噻吩等)时[59], 能与两分子非末端炔烃构建含酰胺键的多元芳香环化合物(Scheme 24).值得一提的是, 两分子炔烃结构相同且是芳基炔烃时, 可直接通过一锅法一步完成反应; 两分子炔烃结构不同且有其中一分子为芳基炔烃时, 可先在酸的作用下完成与芳基炔烃的单分子环化, 再在氧化剂的作用下完成与第二分子炔烃的环化.该反应底物适用范围广, 官能团容忍性好, 可以扩大到克级反应, 适用于工业生产应用.
2015年, Jeganmohan课题组[60]以[(p-cymene)2-RuCl2]2为催化剂实现了NH-甲基苯基亚氨基砜双邻位的芳基化反应, 该反应以苯硼酸作为芳基源, 芳基化后的产物在Pd(OAc)2/PhI(OAc)2的催化氧化体系中进一步发生分子内脱氢偶联生成1, 2-吩噻嗪类化合物(Scheme 25).该反应操作简单, 底物适用范围广, 手性的NH-亚氨基砜也能高收率、高对映选择性地转化成手性环化产物.
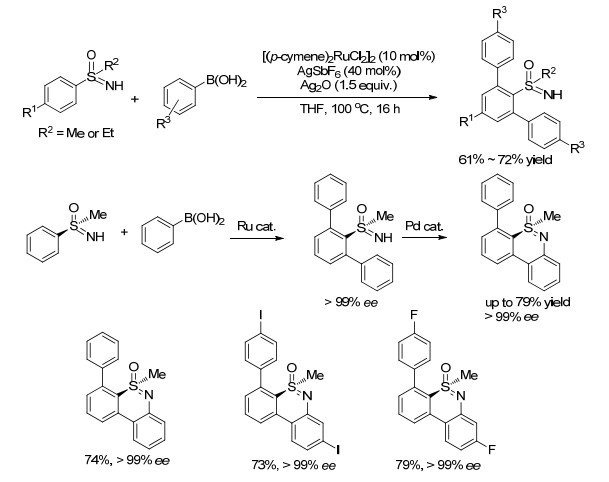
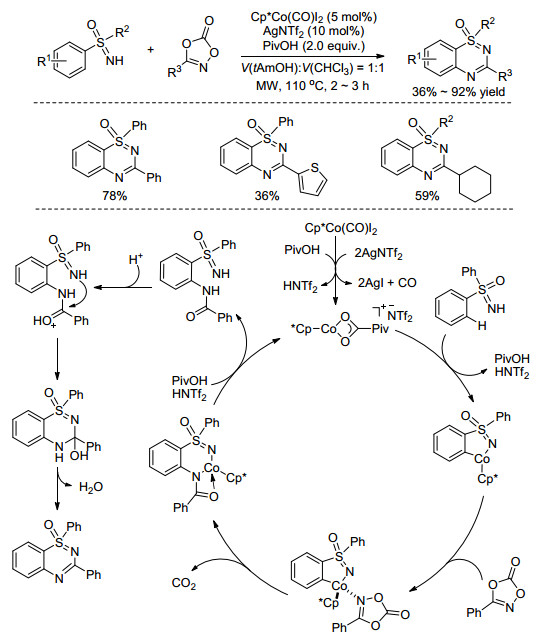
最近, 我们课题组[61]首次利用金属Co作催化剂, 1, 4, 2-二噁唑-5-酮作胺化试剂, 在微波条件下一锅法实现了与NH-亚氨基砜的C—H/N—H的双胺化环化反应, 高效构建苯并噻二嗪-1氧化物(Scheme 26).溶剂对该反应的影响很大, 质子性溶剂有助于反应的顺利进行, 当使用叔戊醇和氯仿的体积比为1:1的混合溶剂时, 产率会大大提升.通过相关的机理研究我们提出了可能的催化循环过程, 首先, 负离子交换形成的活泼金属物种与底物NH-亚氨基砜发生亲电金属化形成环钴中间体, 1, 4, 2-二噁唑-5-酮与之配位, 紧接着脱去CO2、发生酰胺键的插入, 然后去金属化得到邻位酰胺化中间体, 最后在酸性条件下脱氢环化生成最终产物(Scheme 26).
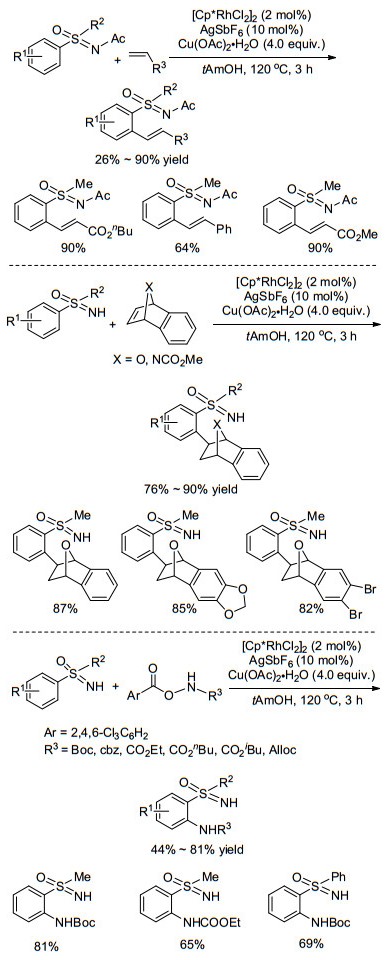
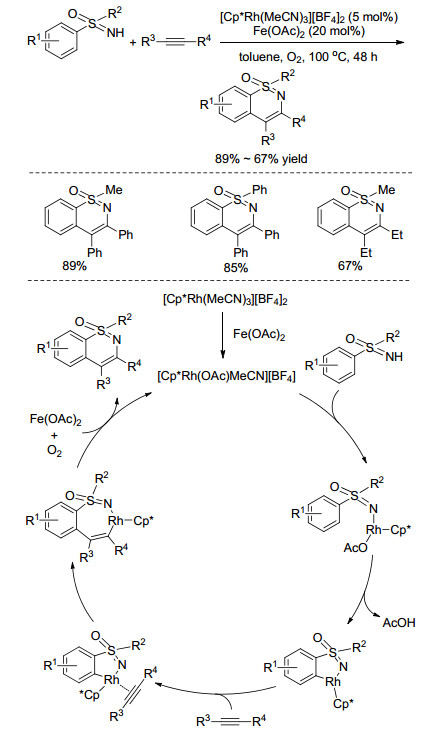
利用过渡金属铑催化亚氨基砜基导向的C(sp2)-H活化, 可以在与亚氨基砜基相连的芳基邻位引入烯基、酰基、胺基、芳基、卤素等功能团, 为构建多样性亚氨基砜提供了有效途径. 2014年, Bolm课题组[62, 63]分别以丙烯酸酯和桥环烯烃为原料, 先后实现了亚氨基砜芳环上的烯基化反应和桥环烷基化反应(Scheme 27, a, b), 最近, 该课题组[64]报道了NH-亚氨基砜与氨基甲酸叔丁酯类化合物在Rh(Ⅲ)的催化下合成2-胺基亚氨基砜, 所得产物可以作为中间体进一步转化为药物分子噻二嗪-1-氧化物(Scheme 27, c).
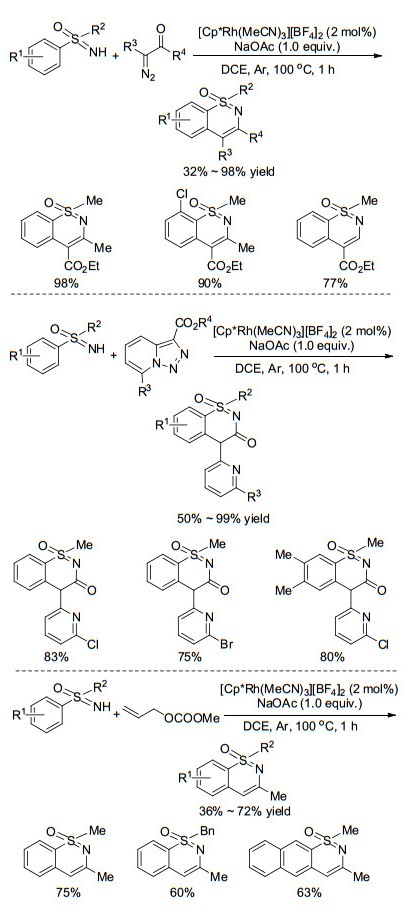
近年来, 亚氨基砜基团除了作为导向基团参与到邻位C-H活化中, 还可以作为偶联体参与到环化反应当中. 2013年, Dong等[65]报道了Rh(Ⅲ)催化的NH-亚氨基砜与二苯乙炔类化合物的胺环反应, 简单高效地构建1, 2-吩噻嗪类衍生物.其反应机理大致为, 导向基团中的氮原子与Rh催化剂配位, 接着发生邻位C(sp2)—H活化得到环铑中间体, 然后与炔烃配位插入、最后还原消除生成最终产物, 同时释放Rh催化剂(Scheme 28).
此后, 该课题组[66]利用重氮化合物作为偶联试剂, 通过卡宾迁移反应与NH-亚氨基砜类化合物一锅法合成1, 2-吩噻嗪类衍生物, 该反应的优点是大大缩短了反应时间, 仅需1 h便可快速完成转化(Scheme 29). 2016年, Lee课题组[67]发展了一种用吡啶并三氮唑作为卡宾前体, 以较高收率合成得到含羰基的1, 2-吩噻嗪类化合物的方法(Scheme 29).作者在反应体系中同时加入等物质的量的的吡啶并三氮唑和重氮化合物, 发现含羰基的产物占多数, 结果表明吡啶并三氮唑比重氮化合物更活泼.最近, Bolm课题组[68]同样利用NH-亚氨基砜为原料, 与烯丙基碳酸酯类化合物反应, 合成了一系列4号位碳上无取代基的1, 2-吩噻嗪类衍生物(Scheme 29).作者对产物进行了一系列修饰, 合成了各种有潜在应用价值的化合物, 拓展这类化合物在合成中的应用.
综上所述, 基于亚氨基砜化合物良好的生物或药物活性, 近几年来关于该类化合物的合成的方法学研究取得了快速的发展.由于亚氨基砜结构本身的稳定性以及其天然的不对称性, 此类官能团参与的反应对于各类基团的兼容性普遍较好, 即使是手性分子也能以高收率、高对映选择性完成相应的转化, 因此极大丰富了亚氨基砜化合物的多样性.但仍然存在诸多挑战, 如合成的化合物在应用方面存在不足, 对于其特殊的生物学方面的活性需要更详尽的研究.随着对亚氨基砜衍生物研究的深入及在有机合成领域应用的推广, 发展低毒性原料或催化体系的多组分反应, 实现绿色方法合成结构多样的亚氨基砜类化合物将是一个值得探讨的方向.
Bentley, H. R.; McDermott, E. E.; Pace, J.; Whitehead, J. K.; Moran, T. Nature 1950, 165, 150. doi: 10.1038/165150a0
(a) Langner, M. ; Bolm, C. Angew. Chem. , Int. Ed. 2004, 43, 5984.
(b) Shen, X. ; Zhang, W. ; Ni, C. ; Gu, Y. ; Hu, J. J. Am. Chem. Soc. 2012, 134, 16999.
(c) Harmata, M. ; Hong, X. J. Am. Chem. Soc. 2003, 125, 5754.
(d) Koep, S. ; Gais, H. -J. ; Raabe, G. J. Am. Chem. Soc. 2003, 125, 13243.
(e) Moessner, C. ; Bolm, C. Angew. Chem. , Int. Ed. 2005, 44, 7564.
Miller, P.; James, G. W. L. Arch. Int. Pharmacodyn. Ther. 1978, 231, 328. http://lf-oll.s3.amazonaws.com/titles/1991/chaucer-the-complete-works-of-geoffrey-chaucer-vol-3.epub
Park, S. J.; Baars, H.; Mersmann, S.; Buschmann, H.; Baron, J. M.; Amann, P. M.; Czaja, K.; Hollert, H.; Bluhm, K.; Redelstein, R.; Bolm, C. ChemMedChem 2013, 8, 217. doi: 10.1002/cmdc.201200403
Lucking, U.; Jautelat, R.; Kruger, M.; Brumby, T.; Lienau, P.; Schafer, M.; Briem, H.; Schulze, J.; Hillisch, A.; Reichel, A.; Siemeister, G. ChemMedChem 2013, 8, 1021. doi: 10.1002/cmdc.201390025
Satzinger, G. Drug News Perspect. 2001, 14, 197. doi: 10.1358/dnp.2001.14.4.858403
Von Nussbaum, F.; Karthaus, D.; Anlauf, S.; Delbeck, M.; Li, V.; Meibom, D.; Lustig, K.; Schneider, D. WO 2010115548, 2010.
Walker, D.; Zawistoski, M.; McGlynn, M.; Li, J.; Kung, D. W.; Bonnette, P.; Baumann, A.; Buckbinder, L.; Houser, J. A.; Boer, J.; Mistry, A.; Han, S.; Xing, L.; Perez, A. B. Bioorg. Med. Chem. Lett. 2009, 19, 3253. doi: 10.1016/j.bmcl.2009.04.093
Ulrich, L. Angew. Chem., Int. Ed. 2013, 52, 9399. doi: 10.1002/anie.v52.36
(a) Bentley, H. R. ; Whitehead, J. K. J. Chem. Soc. 1952, 1572.
(b) Johnson, C. R. ; Haake, M. ; Schroeck, C. W. J. Am. Chem. Soc. 1970, 92, 6594.
(c) Stoss, P. ; Satzinger, G. Angew. Chem. , Int. Ed. 1971, 10, 76.
(d) Johnson, C. R. ; Schroeck, C. W. J. Am. Chem. Soc. 1973, 95, 7418.
(e) Rynbrandt, R. H. ; Balgoyen, D. P. J. Org. Chem. 1978, 43, 1824.
(f) Brandt, J. ; Gais, H. J. Tetrahedron: Asymmetry 1997, 8, 909.
(a) Tamura, Y. ; Sumoto, K. ; Minamikawa, J. ; Ikeda, M. Tetrahedron Lett. 1972, 4137.
(b) Tamura, Y. ; Minamikawa, J. ; Sumoto, K. ; Fujii, S. ; Ikeda, M. J. Org. Chem. 1973, 38, 1239.
(c) Johnson, C. R. ; Kirchhoff, R. A. ; Corkins, H. G. J. Org. Chem. 1974, 39, 2458.
(d) Allenmark, S. ; Claeson, S. ; Lowendahl, C. Tetrahedron: Asymmetry 1996, 7, 361.
(a) Takada, H. ; Ohe, K. ; Uemura, S. Angew. Chem. , Int. Ed. 1999, 38, 1288.
(b) Lacote, E. ; Amatore, M. ; Fensterbank, L. ; Malacria, M. Synlett 2002, 116.
(c) Cren, S. ; Kinahan, T. C. ; Skinner, C. L. ; Tye, H. Tetrahedron Lett. 2002, 43, 2749.
(d) Okamura, H. ; Bolm, C. Org. Lett. 2004, 6, 1305.
(e) Cho, G. Y. ; Bolm, C. Org. Lett. 2005, 7, 4983.
(f) Mancheno, O. G. ; Bolm, C. Org. Lett. 2006, 8, 2349.
(g) Mancheno, O. G. ; Dallimore, J. ; Plant, A. ; Bolm, C. Org. Lett. 2009, 11, 2429.
Miao, J.; Richards, N. G. J.; Ge, H. Chem. Commun. 2014, 50, 9687. doi: 10.1039/C4CC04349A
Zenzola, M.; Doran, R.; Degennaro, L.; Luisi, R.; Bull, J. A. Angew. Chem., Int. Ed. 2016, 55, 7203. doi: 10.1002/anie.201602320
Wang, J.; Frings, M.; Bolm, C. Chem.-Eur. J. 2014, 20, 966. doi: 10.1002/chem.201303850
Cheng, Y.; Dong, W.; Wang, L.; Parthasarathy, K.; Bolm, C. Org. Lett. 2014, 16, 2000. doi: 10.1021/ol500573f
Teng, F.; Cheng, J.; Yu, J.-T. Org. Biomol. Chem. 2015, 13, 9934. doi: 10.1039/C5OB01558H
Teng, F.; Sun, S.; Jiang, Y.; Yu, J.-T.; Cheng, J. Chem. Commun. 2015, 51, 5902. doi: 10.1039/C5CC00839E
Teng, F.; Cheng, J.; Bolm, C. Org. Lett. 2015, 17, 3166. doi: 10.1021/acs.orglett.5b01537
Wang, H.; Frings, M.; Bolm, C. Org. Lett. 2016, 18, 2431. doi: 10.1021/acs.orglett.6b00958
Dehli, J. R.; Bolm, C. J. Org. Chem. 2004, 69, 8518. doi: 10.1021/jo0485583
Chen, X.-Y.; Bohmann, R. A.; Wang, L.; Dong, S.; R uber, C.; Bolm, C. Chem.-Eur. J. 2015, 21, 10330. doi: 10.1002/chem.201501629
Chen, Z.; Huang, J.; Wang, Z. J. Org. Chem. 2016, 81, 9308. doi: 10.1021/acs.joc.6b01891
Wang, L.; Huang, H.; Priebbenow, D. L.; Pan, F.-F.; Bolm, C. Angew. Chem., Int. Ed. 2013, 52, 3478. doi: 10.1002/anie.v52.12
Priebbenow, D. L.; Becker, P.; Bolm, C. Org. Lett. 2013, 15, 6155. doi: 10.1021/ol403106e
Chen, X. Y.; Wang, L.; Frings, M.; Bolm, C. Org. Lett. 2014, 16, 3796. doi: 10.1021/ol5016898
Wang, H.; Cheng, Y.; Becker, P.; Raabe, G.; Bolm, C. Angew. Chem., Int. Ed. 2016, 55, 12655. doi: 10.1002/anie.201605743
Pirwerdjan, R.; Priebbenow, D. L.; Becker, P.; Lamers, P.; Bolm, C. Org. Lett. 2013, 15, 5397. doi: 10.1021/ol4026028
Pirwerdjan, R.; Becker, P.; Bolm, C. Org. Lett. 2015, 17, 5008. doi: 10.1021/acs.orglett.5b02477
Pirwerdjan, R.; Becker, P.; Bolm, C. Org. Lett. 2016, 18, 3307. doi: 10.1021/acs.orglett.6b01646
Bolm, C.; Hildebrand, J. P. J. Org. Chem. 2000, 65, 169. doi: 10.1021/jo991342u
Cho, G. Y.; Rémy, P.; Jansson, J.; Moessner, C.; Bolm, C. Org. Lett. 2004, 6, 3293. doi: 10.1021/ol048806h
Sedelmeier, J.; Bolm, C. J. Org. Chem. 2005, 70, 6904. doi: 10.1021/jo051066l
Moessner, C.; Bolm, C. Org. Lett. 2005, 7, 2667. doi: 10.1021/ol050816a
Kim, J.; Ok, J.; Kim, S.; Choi, W.; Lee, P. H. Org. Lett. 2014, 16, 4602. doi: 10.1021/ol502174n
Miyasaka, M.; Hirano, K.; Satoh, T.; Kowalczyk, R.; Bolm, C.; Miura, M. Org. Lett. 2010, 13, 359. doi: 10.3998/ark.5550190.0013.611
Wang, L.; Priebbenow, D. L.; Dong, W.; Bolm, C. Org. Lett. 2014, 16, 2661. doi: 10.1021/ol500963p
Aithagani, S. K.; Kumar, M.; Yadav, M.; Vishwakarma, R. A.; Singh, P. P. J. Org. Chem. 2016, 81, 5886. doi: 10.1021/acs.joc.6b00593
Aithagani, S. K.; Dara, S.; Munagala, G.; Aruri, H.; Yadav, M.; Singh, P. P. Org. Lett. 2015, 17, 5547. doi: 10.1021/acs.orglett.5b02804
Priebbenow, D. L.; Bolm, C. Org. Lett. 2014, 16, 1650. doi: 10.1021/ol5003016
Zou, Y.; Xiao, J.; Peng, Z.; Dong, W.; An, D. Chem. Commun. 2015, 51, 14889. doi: 10.1039/C5CC05483D
Qin, W. J.; Li, Y.; Yu, X.; Deng, W.-P. Tetrahedron 2015, 71, 1182. doi: 10.1016/j.tet.2015.01.013
Dong, S.; Frings, M.; Cheng, H.; Wen, J.; Zhang, D.; Raabe, G.; Bolm, C. J. Am. Chem. Soc. 2016, 138, 2166. doi: 10.1021/jacs.6b00143
Zhou, H.; Hong, J.; Huang, J.-P.; Chen, Z.-Y. Asian J. Org. Chem. 2017, 6, 817. doi: 10.1002/ajoc.v6.7
Teng, F.; Yu, J. T.; Jiang, Y.; Yang, H.; Cheng, J. Chem. Commun. 2014, 50, 8412. doi: 10.1039/c4cc03439b
Hu, W.; Teng, F.; Peng, H.; Yu, J.; Sun, S.; Cheng, J. Shao, Y. Tetrahedron Lett. 2015, 56, 7056. doi: 10.1016/j.tetlet.2015.11.025
Teng, F.; Yu, J.-T.; Zhou, Z.; Chu, H.; Cheng, J. J. Org. Chem. 2015, 80, 2822. doi: 10.1021/jo502607c
Dannenberg, C. A.; Fritze, L.; Krauskopf, F.; Bolm, C. Org. Biomol. Chem. 2017, 15, 1086. doi: 10.1039/C6OB02691E
Peng, H.; Yu, J. T.; Bao, W.; Xu, J.; Cheng, J. Org. Biomol. Chem. 2015, 13, 10600. doi: 10.1039/C5OB01705J
Bohnen, C.; Bolm, C. Org. Lett. 2015, 17, 3011. doi: 10.1021/acs.orglett.5b01384
Zhu, H.; Yu, J.-T.; Cheng, J. Chem. Commun. 2016, 52, 11908. doi: 10.1039/C6CC06359D
Yadav, M. R.; Rit, R. K.; Sahoo, A. K. Chem.-Eur. J. 2012, 18, 5541. doi: 10.1002/chem.v18.18
Rit, R. K.; Yadav, M. R.; Sahoo, A. K. Org. Lett. 2012, 14, 3724. doi: 10.1021/ol301579q
Rit, R. K.; Yadav, M. R.; Ghosh, K.; Shankar, M.; Sahoo, A. K. Org. Lett. 2014, 16, 5258. doi: 10.1021/ol502337b
Yadav, M. R.; Rit, R. K.; Sahoo, A. K. Org. Lett. 2013, 15, 1638. doi: 10.1021/ol400411v
Yadav, M. R.; Rit, R. K.; Shankar, M.; Sahoo, A. K. J. Org. Chem. 2014, 79, 6123. doi: 10.1021/jo5008465
Yadav, M. R.; Shankar, M.; Ramesh, E.; Ghosh, K.; Sahoo, A. K. Org. Lett. 2015, 17, 1886. doi: 10.1021/acs.orglett.5b00570
Raghuvanshi, K.; Zell, D.; Ackermann, L. Org. Lett. 2017, 19, 1278. doi: 10.1021/acs.orglett.6b03898
Shankar, M.; Ghosh, K.; Mukherjee, K.; Rit, R. K.; Sahoo, A. K. Org. Lett. 2016, 18, 6416. doi: 10.1021/acs.orglett.6b03314
Chinnagolla, R. K.; Vijeta, A.; Jeganmohan, M. Chem. Commun. 2015, 51, 12992. doi: 10.1039/C5CC04589D
Huang, J.; Huang, Y.; Wang, T.; Huang, Q.; Wang, Z.; Chen, Z.-Y. Org. Lett. 2017, 19, 1128. doi: 10.1021/acs.orglett.7b00120
Parthasarathy, K.; Bolm, C. Chem.-Eur. J. 2014, 20, 4896. doi: 10.1002/chem.201304925
Dong, W.; Parthasarathy, K.; Cheng, Y.; Pan, F.; Bolm, C. Chem.-Eur. J. 2014, 20, 15732. doi: 10.1002/chem.v20.48
Cheng, Y.; Dong, W.; Wang, H.; Bolm, C. Chem.-Eur. J. 2016, 22, 10821. doi: 10.1002/chem.201602550
Dong, W.; Wang, L.; Parthasarathy, K.; Pan, F.; Bolm, C. Angew. Chem., Int. Ed. 2013, 52, 11573. doi: 10.1002/anie.201304456
Cheng, Y.; Bolm, C. Angew. Chem., Int. Ed. 2015, 127, 12526. doi: 10.1002/ange.201501583
Jeon, W. H.; Son, J. Y.; Kim, J. E.; Lee, P. H. Org. Lett. 2016, 18, 3498. doi: 10.1021/acs.orglett.6b01750
Wen, J.; Tiwari, D. P.; Bolm, C. Org. Lett. 2017, 19, 1706. doi: 10.1021/acs.orglett.7b00488

图 1 一些含有亚氨基砜骨架的药物分子
Figure 1 Some pharmaceutical molecules containing sulfoximine core structure

Scheme 1 从亚砜合成NH-亚氨基砜的方法
Scheme 1 Strategies for the preparation of NH-sulfoximines from sulfoxides

Scheme 2 光学活性亚氨基砜的合成方法
Scheme 2 Strategies for the preparation of optically active sulfoximines

Scheme 3 铁催化亚氨基砜与二芳基甲烷的氧化偶联反应
Scheme 3 Iron-catalyzed oxidative coupling reactions of sulfoximines with diarylmethanes

Scheme 5 亚氨基砜的N-CF3化反应及自身环化反应
Scheme 5 N-CF3 reactions and self-cyclizations of sulfoximines

Scheme 6 钯催化亚氨基砜与烯烃的N-烯基化反应
Scheme 6 Palladium-catalyzed N-vinylations of sulfoximines with olefins

Scheme 7 Ru催化N-烯胺基亚氨基砜的氧化反应
Scheme 7 Ruthenium-catalyzed oxidative reactions of N-enamino sulfoximines

Scheme 9 高碘试剂参与的亚氨基砜N-炔基化反应
Scheme 9 Iodine reagent involved N-alkynylations of sulfoximines

Scheme 10 无金属催化N-炔基亚氨基砜的转化反应
Scheme 10 Metal-free catalyzed transformations of N-alkynylated sulfoximines

Scheme 11 铜催化亚氨基砜与卤代芳烃的N-芳化反应
Scheme 11 Copper-catalyzed N-arylations of sulfoximines with aryl halides

Scheme 12 铜催化亚氨基砜与芳基硼酸、芳基硅烷的N-芳化反应
Scheme 12 Copper-catalyzed N-arylations of sulfoximines with aryl boronic acids or aryl siloxanes

Scheme 13 铜催化亚氨基砜与杂芳烃的N-芳化反应
Scheme 13 Copper-catalyzed N-arylations of sulfoximines with heteroarenes

Scheme 14 亚氨基砜与吖嗪氮氧化合物、芳炔前体的N-芳化反应
Scheme 14 N-Arylations of sulfoximines with azine N-oxides or aryne precuesors

Scheme 15 亚氨基砜与甲基芳烃的N-芳酰化反应
Scheme 15 N-aroylations of sulfoximines with methyl arenes

Scheme 19 铜催化亚氨基砜与硅烷的N-硅化反应
Scheme 19 Copper-catalyzed N-silylations of sulfoximines with silanes

Scheme 20 过渡金属催化亚氨基砜与其它偶联试剂的N-官能化反应
Scheme 20 Transition metal-catalyzed N-functionalization of sulfoximines with other coupling reagents

Scheme 21 钯催化亚氨基砜的C(sp2)—H键官能化反应
Scheme 21 Palladium-catalyzed C(sp2)—H bond functionalization of sulfoximines

Scheme 22 钯催化亚氨基砜的C(sp3)—H键官能化反应
Scheme 22 Palladium-catalyzed C(sp3)—H bond functionalization of sulfoximines

Scheme 23 钌催化亚氨基砜基导向的芳基邻位C—H键官能化反应
Scheme 23 Ruthenium-catalyzed sulfoximine-directed ortho C—H functionalization of arenes

Scheme 24 钌催化亚氨基砜基导向的杂环芳烃双环化反应
Scheme 24 Ruthenium-catalyzed sulfoximine-directed diannulation of heteroarenes

Scheme 25 钌催化亚氨基砜与芳基硼酸的双邻位芳基化反应
Scheme 25 Ruthenium-catalyzed di-ortho arylation of sulfoximines with aromatic boronic acids

Scheme 26 钴催化亚氨基砜与1, 4, 2-二噁唑-5-酮的胺化环化反应
Scheme 26 Cobalt-catalyzed amination/cyclization of sulfoximines with 1, 4, 2-dioxazol-5-ones

Scheme 27 铑催化亚氨基砜基导向的芳基邻位C—H键官能化反应
Scheme 27 Rhodium-catalyzed sulfoximine-directed ortho C—H functionalization of arenes

Scheme 28 铑催化亚氨基砜与炔烃的氧化环化反应
Scheme 28 Rhodium-catalyzed oxidative annulation of sulfoximines with alkynes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:





