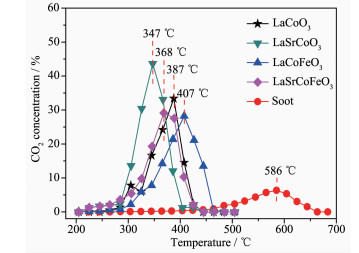
图 1.
催化氧化碳烟活性曲线
Figure 1.
Catalytic oxidation activity curve of soot

柴油机以其良好的燃料利用率、耐久性和CO2排量低等优点,广泛应用于轮船、汽车、货车、发电厂等大型设备[1]。然而,由于油品燃烧不完全导致柴油机尾气中含有大量碳烟(soot)颗粒,对环境和人体健康造成较大危害[2-3]。随着国家对移动源尾气排放法规的日趋严格,仅机内净化技术已难以达到当前的排放标准,因此,开发一种性能优良的柴油机尾气净化技术显得势在必行。目前针对尾气中碳烟去除主要安装堇青石DPF(颗粒过滤器)过滤[4],但随着碳烟在DPF壁内积累易导致发动机排气背压增加,使柴油机运行工况恶化并产生更多污染物,降低其运行寿命,而碳烟在发动机(200~400 ℃)中极难自身氧化分解,因此针对这一难题研究一种催化氧化碳烟的高效催化剂显得势在必行。另一方面,由于当前原油炼制中脱硫的不完全性导致柴油机尾气中含有一定SO2,易使催化剂中毒失活,且有关催化剂抗硫性能和中毒机理亦是当前环境催化领域一大热点。因此,研究催化氧化碳烟性能和抗硫中毒机理对设计、指导高效催化剂开发具有较大的理论和实际意义。
目前在各类移动源尾气催化剂中,主要有金属氧化物[5]、贵金属[6]、尖晶石[7]和钙钛矿型[8]催化剂。其中钙钛矿催化剂相较其它催化剂具有价格便宜、热稳定性好,催化活性高等优点,被广泛应用于环境催化领域[9-10]。钙钛矿命名来源于稀土金属氧化物CaTiO3,其分子式为ABO3[11];通过金属离子掺杂可使催化剂性能得到较好调变,通常钙钛矿A位可被碱金属或碱土金属取代,B位可被过渡金属取代而保持其晶体结构不变;其中,碱土金属掺杂对钙钛矿催化剂性能调变至关重要。Dai等[12]通过模板法制备Sr掺杂的Eu1-xSrxFeO3催化剂,研究表明Sr掺杂能增加催化剂表面吸附氧浓度和低温还原性能;Liu等[13]采用共沉淀法制备La1-xSrxMnO3,通过SEM表征发现Sr掺杂能形成超细纳米颗粒,对催化氧化CH4表现出较高催化活性;Gao等[14]制备纳米钙钛矿催化剂La1-xSrxTiO3,并用于催化氧化碳烟具有较好的催化活性,表明金属Sr掺杂钙钛矿催化剂具有较好的性能调变潜力。另一方面,Fe基氧化物催化剂在环境催化领域也有广泛应用,如FeTiOx[15]、Fe2O3-SiO2[16]、WOx/Fe2O3[17];Li等[18]利用柠檬酸配位法制备了La0.9K0.1Co1-xFexO3,研究表明Fe掺杂能提高晶格氧活性,极大促进对碳烟的催化氧化,这表明Fe调变催化剂性能有较好应用前景。催化剂抗硫性能也是工业尾气催化剂的重要指标,研究表明SO2易使催化剂中毒失活,且有关抗硫机理也是当前热点。Jin等[19]制备了Mn/TiO2和Mn-Ce/TiO2催化剂,研究表明当SO2引入Mn/TiO2时其NO转化率从90%降低至30%;Yu等[20]制备了PrOx(0.2)-MnOx/SAPO-34催化剂,研究表明当SO2气氛出现时NO转化率降低81.0%;Li等[21]制备了CeO2-MnOx催化剂,研究表明当通入SO2时NO转化率降低至75%,即使停止通入SO2时其催化活性也未能恢复,表明SO2对催化剂活性有较大影响,且较难活化恢复。Liu等[22]利用Sm掺杂CeO2-TiO2催化剂,通过XPS和DFT (密度泛函理论)计算表明,SmCeTi催化剂抗硫性来源于Sm2++Ce4+?葑Sm3++Ce3+氧化还原反应,有效地抑制了电子从SO2向Ce4+迁移,使催化剂具有较好的抗硫能力;Jiang等[23]制备了V2O5共掺杂Mn-Ce(0.4)-V/AC催化剂,研究表明催化剂抗硫性来源于V2O5掺杂形成的酸性和Mn-Ce固溶体的分散,有效抑制了SO2对Mn-Ce的硫酸化;Yu等[24]制备的MxCe1-xOδ/3DOM-mSiO2催化剂表现出良好的催化燃烧碳烟活性,通过XRD、Raman、DFT等证明,SO2中毒主要由于催化剂吸附SO2与活性氧及金属原子反应形成硫酸盐物种,导致催化活性降低,这表明SO2对催化剂活性有较大影响,且有关催化剂抗硫性能优化和机理也未十分清晰,这也是当前催化剂抗硫研究一大热点。因此,针对催化剂性能优化及抗硫机理研究对未来移动源尾气净化具有较好借鉴意义。
本工作采用柠檬酸-EDTA配位法制备4种性能较佳的钙钛矿催化剂La1-xSrxCo1-yFeyO3(x=0,0.1;y=0,0.3),分别研究金属Sr、Fe掺杂对催化剂物理化学性能调变和抗硫中毒机理,以期对未来机动车尾气催化剂的设计、开发提供有效理论支撑。
通过柠檬酸-EDTA配位法制备钙钛矿催化剂La1-xSrxCo1-yFeyO3,分别记为LaCoO3、La0.9Sr0.1CoO3(LaSrCoO3)、LaCo0.7Fe0.3O3(LaCoFeO3)、La0.9Sr0.1Co0.7Fe0.3O3(LaSrCoFeO3)。即按化学计量比称取La(NO3)3·6H2O、Sr(NO3)2、Co(NO3)2·6H2O、Fe(NO3)3·9H2O、柠檬酸和EDTA,保持总金属离子、柠檬酸、EDTA的物质的量之比为1:1:0.2,并用20 mL去离子水溶解使上述金属硝酸盐充分混合。将混合溶液置于80 ℃恒温水浴锅中加热搅拌,至溶液逐渐形成粘稠状凝胶;后转移至150 ℃恒温油浴中保温2 h使凝胶充分膨胀得到疏松多孔的干凝胶,再转移至120 ℃恒温干燥箱中干燥过夜得催化剂前驱体。经破碎、研磨转移至真空管式炉中,在空气气氛下以5 ℃·min-1升温至400 ℃焙烧2 h,再升温至750 ℃焙烧5 h,得黑色催化剂粉体。将上述催化剂于300 ℃下在以N2为平衡气的0.08%(V/V)SO2中硫化处理2 h,空速(GHSV)为10 000 h-1[25]。硫化后的催化剂分别记为LaCoO3-S、LaSrCoO3-S、LaCoFeO3-S、LaSrCoFeO3-S。使用后的催化剂分别记为LaCoO3-U、LaSrCoO3-U、LaCoFeO3-U、LaSrCoFeO3-U。
催化剂的XRD分析使用荷兰PANalytical公司生产的PW3040/60型X射线衍射仪。采用Co Kα(λ=0.179 03 nm)辐射源,管电压40 kV,管流40 mA,衍射角2θ=10°~90°,扫描步长0.02°。
傅利叶红外光谱测试(FT-IR)在Thermo Nicolet公司生产的Nexus FT-IR红外光谱仪上进行。光谱分辨率4 cm-1,扫描范围为4 000~400 cm-1。催化剂样品与KBr按质量比1:100混合并充分干燥研磨并压片测试。
采用日本JEOR公司生产的JSM-7001F型热场发射扫描电子显微镜对催化剂表面形貌进行SEM分析。测试前对样品进行喷金处理以增强其导电性,扫描电镜工作电压为10 kV。采用能量分散谱仪(EDS)分析样品的元素组成。
程序升温还原使用天津先权公司TP 5080型催化剂评价装置,50 mg催化剂在N2气氛下从室温以10 ℃·min-1程序升温至400 ℃保持1 h,后降至室温混氢30 min待稳定基线,气氛为95% N2和5% H2(V/V),升温速率为30 ℃·min-1。程序升温至900 ℃,使用TCD检测器记录,水气使用干燥分子筛吸附。
催化剂的元素价态扫描在Thermo ESCALAB 250Xi型X射线光电子能谱仪(XPS)上进行,采用Al Kα(hν=1 486.6 eV)作为光源, 真空度为5×10-8 Pa,以污染碳(C1s:284.8 eV)用于校准作为能量标准,结合能误差为±0.2 eV。采用Gaussian-Lorentzian法及Shirley扣除背景,计算表面原子含量和价态分布。
程序升温脱附使用天津先权公司TP 5080型催化剂评价装置,取150 mg催化剂在He气氛下从室温以10 ℃·min-1程序升温至400 ℃保持1 h,后降至室温切换3%(V/V)SO2气氛吸附30 min,再切换为He气充分吹扫表面物理吸附的SO2至基线稳定。在30 mL·min-1纯He气氛中程序升温至900 ℃,使用TCD检测器记录信号。
实验使用PerkinElmer公司生产的TGA 4000型热重分析仪,取30 mg催化剂在N2气氛下从室温以10 ℃·min-1程序升温至800 ℃。
采用天津先权公司生产的连续气固相反应装置(WFS-3015)评价催化氧化碳烟活性。采用紧密接触以克服反应中不均匀传热和传质,按1:9质量比称取碳烟和催化剂,充分研磨并搅拌,置入干燥箱干燥,以保持各催化剂具有相同接触条件,碳烟选用Degussa公司生产的Printex-U碳代替柴油机尾气中碳烟颗粒。反应使用180 mg催化剂,气氛为0.15%(V/V)NO+5%(V/V)O2+N2平衡气,混合气总流量为100 mL·min-1,反应空速(GHSV)为15 000 h-1,在温度为200~500 ℃内评价其催化氧化碳烟活性。碳烟氧化产生的CO2及气氛中O2的浓度采用安捷伦科技有限公司生产的GC 7820A型气相色谱检测。本工作中,碳烟开始燃烧温度定义为起燃温度(Ti),碳烟最大燃烧速率时温度定义为(Tm),碳烟燃尽温度定义为(Tf)。式(1)为碳烟燃烧产生的CO2在气氛中所占的浓度(CCO2),该浓度越大表明催化燃烧碳烟速率越快,一般用于评价催化氧化碳烟活性;其Ti、Tm和Tf越低,表明催化剂活性越高。
|
$ \mathrm{CO}_{2} \text { concentration }(\%)=\frac{\alpha_{\mathrm{CO}_{2}}}{\alpha_{\mathrm{CO}_{2}}+\alpha_{\mathrm{O}_{2}}} \times 100 \% $ |
(1) |
其中,αCO2为反应器出口气体中的CO2浓度;αO2为反应器出口气体中的O2浓度。
图 1为催化剂催化氧化碳烟活性曲线,碳烟燃烧在无催化剂时Tm为586 ℃,在柴油机尾气中(200~400 ℃)极难自身氧化分解。当反应引入LaCoO3催化剂时,碳烟催化氧化温度降低33.8%,其Tm为387 ℃; 当进一步掺杂金属Sr后, 发现LaSrCoO3具有较高的催化燃烧碳烟活性(碳烟氧化温度降低40.6%),其Tm仅为347 ℃,表明Sr掺杂有利于提高对碳烟催化活性。当催化剂同时掺杂Sr、Fe时,催化氧化碳烟活性并未进一步提高(Tm为368 ℃),表明Sr、Fe掺杂抑制了催化氧化碳烟活性提高。另一方面,催化剂抗硫性能也是重要评价指标。表 1表明催化剂经过SO2硫化后,SO2对LaCoO3活性有较大影响(Tm升高24.5%),其Tm为482 ℃;而当LaCoO3同时掺杂Sr、Fe后,其催化氧化碳烟活性(Ti=320 ℃,Tm=361 ℃,Tf=460 ℃)变化较小,表明同时掺杂Sr、Fe有利于改善LaCoO3抗硫中毒能力。综上所述,采用柠檬酸-EDTA配位法制备的催化剂具有较好的催化氧化碳烟活性,其中Sr掺杂有较好的催化碳烟活性;同时掺杂Sr、Fe虽未使活性进一步提高,但有利于提高其抗硫性能。
 下载:
导出CSV
下载:
导出CSV
| Sample | Ti / ℃ | Tm / ℃ | Tf / ℃ |
| LaCoO3 | 312 | 387 | 426 |
| LaSrCoO3 | 284 | 347 | 404 |
| LaCoFeO3 | 323 | 407 | 475 |
| LaSrCoFeO3 | 307 | 368 | 421 |
| LaCoO3-S | 421 | 482 | 500 |
| LaSrCoO3-S | 321 | 381 | 461 |
| LaSrCoFeO3-S | 320 | 361 | 460 |
| Soot | 400 | 586 | 675 |
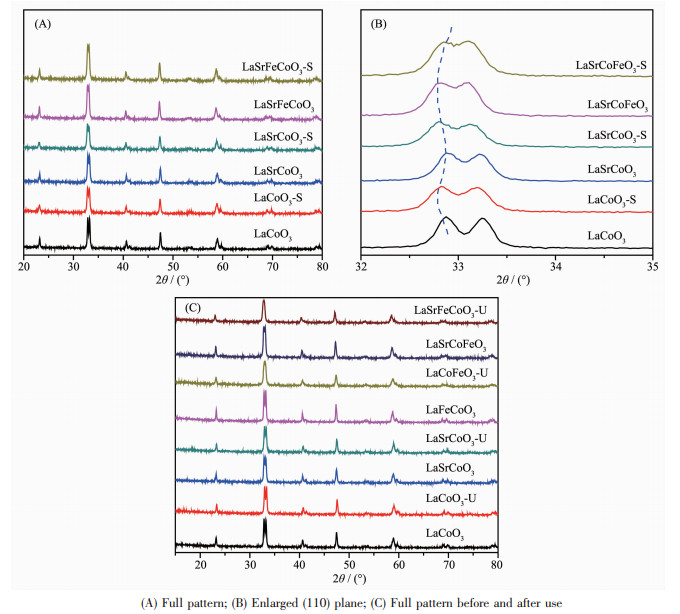
为研究金属掺杂和SO2对催化剂结构影响,对硫化前后的La1-xSrxCo1-yFeyO3催化剂进行XRD表征。图 2(A)表明各催化剂主要特征衍射峰与LaCoO3(PDF No.75-0279)标准卡一致,表明LaCoO3催化剂掺杂Sr、Fe后仍保持完整钙钛矿结构。值得注意的是,La1-xSrxCo1-yFeyO3-S的特征峰与新鲜催化剂基本一致,无明显硫酸盐衍射峰出现,表明硫化后对钙钛矿晶型无明显破坏。由图 2(B)可知,催化剂掺杂Sr、Fe后,衍射峰向低角度偏移。根据Bragg′s Law,由于Sr2+(0.118 nm)、Fe3+(0.065 4 nm)离子半径大于La3+(0.103 2 nm)、Co3+(0.061 nm),使Sr、Fe能成功进入钙钛矿晶格结构。而相较新鲜催化剂,硫化后衍射峰进一步向低角度偏移,这可能由于SO2与掺杂金属或表面吸附氧结合而引起的晶格膨胀;当催化剂同时掺杂Sr、Fe时,La1-xSrxCo1-yFeyO3-S衍射峰向高角度偏移,这可能由于SO2与掺杂金属结合导致的晶格收缩。图 2(C)为使用前后催化剂XRD图对比,使用前后催化剂晶型结构无明显变化,表明各催化剂具有较好结构稳定性。
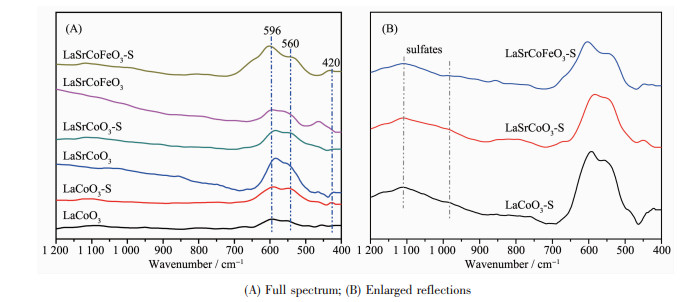
为进一步研究金属掺杂对La1-xSrxCo1-yFeyO3催化剂的结构影响,对各催化剂的FT-IR图谱进行研究(图 3)。钙钛矿ABO3基本结构为BO6八面体,八面体之间的间隙原子是A位阳离子,BO6红外谱图有6种振动模式。如果3对B-O键具有相同键长,即BO6八面体具有高对称性时,伸缩振动(ν3)是非红外活性的;当BO6八面体对称性较低时,B-O键伸缩振动(ν3)则具有红外活性[26]。由图可知,所有样品主要存在3种红外振动,分别在596、560和420 cm-1处。研究表明波数在596和560 cm-1处振动峰归属于CoO6八面体中的2种Co-O伸缩振动,而420 cm-1处的振动归属于CoO6中Co-O键的弯曲振动[9];该结果进一步证明制备的La1-xSrxCo1-yFeyO3催化剂样品具有明显钙钛矿型氧化物(ABO3)结构。此外,由图 3(B)可知,催化剂硫化后在975、1 074、1 137 cm-1处形成明显混合峰,这主要归属为硫酸盐物种(SO32-和SO42-)在催化剂表面的沉积[27]。综上所述,Sr、Fe掺杂仍能保持完整钙钛矿结构,且SO2对钙钛矿结构无明显影响,SO2主要以硫酸盐形式存在。
为研究金属掺杂及SO2对催化剂微观形貌影响,对La1-xSrxCo1-yFeyO3催化剂进行SEM表征。LaCoO3颗粒形状大小不均且出现一定程度团聚;当样品掺杂Sr后,其颗粒分散度较好,无明显烧结现象,且颗粒呈球形分布,尺寸大小在110 nm左右,其催化剂颗粒尺寸与真实碳烟颗粒(70~100 nm)处于同一量级[28],这使得两者可达到较大接触面积,为反应提供必要气-固-固接触位点。而同时掺杂Sr、Fe时,颗粒尺寸有一定变大趋势,这可能是导致催化氧化碳烟活性下降原因之一。当催化剂硫化后,LaCoO3-S颗粒进一步团聚在一起,且颗粒之间呈无规则堆叠,这不利于催化剂与气相接触, 而LaSrCoO3-S、LaSrCoFeO3-S与新鲜催化剂相比,颗粒分布和尺寸无明显变化,表明SO2对催化剂表面颗粒状态及形貌没有明显破坏,故Sr、Fe掺杂提高了催化剂对SO2的耐受性。图 5为催化剂硫化后的EDS-mapping照片,发现硫化后催化剂LaSrCoFeO3-S中S原子百分含量少于LaCoO3催化剂(XPS将进一步确定S的相对含量),表明Sr、Fe掺杂有利于抑制SO2在催化剂表面沉积,降低SO2对催化剂毒化;其它各元素(La、Co、O、Sr、Fe)在催化剂中亦有较好分布,表明元素间可能存在一定相互作用,有利于催化活性提高和抗硫性能改善。

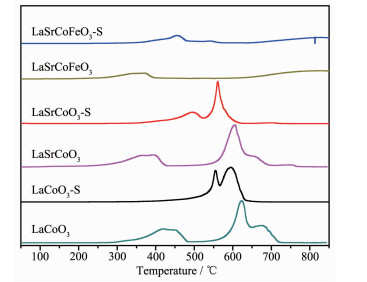
为研究金属掺杂和SO2对催化剂氧化还原性能影响,测定了La1-xSrxCo1-yFeyO3催化剂H2-TPR谱图(图 6)。研究表明H2不仅能还原催化剂中变价金属离子,其表面氧物种亦能参与还原反应,这在一定程度上可反映催化剂中表面氧物种活性[29]。LaCoO3有2个主要的还原峰,在440 ℃处还原峰归属为Co3+还原为Co2+和表面吸附氧物种,622 ℃处还原峰归属为Co2+还原为Co和晶格氧物种[1]。当Sr掺杂LaCoO3后还原峰向低温偏移,表明Sr掺杂有利于提高催化剂低温还原性能和表面氧物种活性。当催化剂同时掺杂Sr、Fe时,其高温还原峰消失,低温还原峰并向左偏移,表明Sr、Fe存在较强的相互作用,这导致催化剂中价态环境变化,有利于增加低温还原性能和表面氧物种活性。La1-xSrxCo1-yFeyO3硫化后的还原性能有一定程度的降低,其中硫化对LaCoO3还原性能影响较大(低温还原峰温升高130 ℃),表明SO2可能减少催化剂中Co2+/Co3+及表面氧物种含量;而LaSrCoFeO3-S低温还原性能虽有一定降低(低温还原峰温为455 ℃),但低温还原性能仍优于LaSrCoO3(低温还原峰温500 ℃)和LaCoO3 (低温还原峰温为556 ℃),表明Sr、Fe同时掺杂对低温还原性能和抗硫性能有较大改善。综上所述,催化剂低温还原性能和抗硫能力顺序为:LaSrCoFeO3 > LaSrCoO3 > LaCoO3,催化剂中毒可能主要来源于SO2对Co2+/Co3+和表面吸附氧的硫酸化。
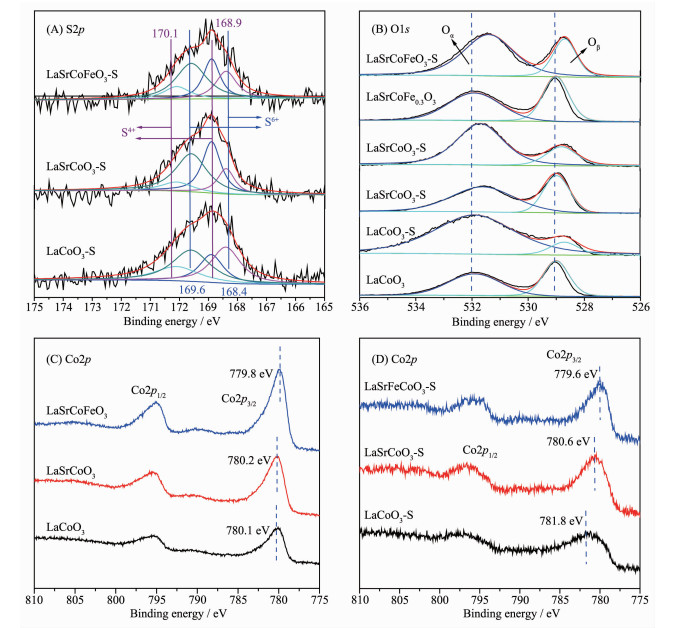
为进一步研究催化剂中硫物种价态及各元素化学环境,研究了S2p的XPS价态分布(图 7(A))。研究表明,168.4和169.6 eV处的谱峰分别归属为S6+的2p3/2和2p1/2;在~168.9和170.1 eV处分别归属为S4+的2p3/2和2p1/2[30-31]。当La1-xSrxCo1-yFeyO3硫化后,S2p的分峰拟合图表明催化剂中SO2主要以SO32-和SO42-形式存在,这也与FT-IR结果一致。表 2表明LaSrCoFeO3-S表面硫的原子百分含量(3.18%)少于LaSrCoO3(4.00%)和LaCoO3(4.06%),表明同时掺杂Sr、Fe有利于抑制SO2在催化剂表面沉积,这也与EDS-mapping结果一致。综上所述,同时掺杂Sr、Fe有利于减少SO2在催化剂表面的沉积,且SO2在催化剂上以硫酸盐(SO32-和SO42-)形式存在。
 下载:
导出CSV
下载:
导出CSV
| Sample | La | Co | O | S | Sr | Fe | C |
| LaCoO3 | 53.21 | 12.23 | 18.81 | — | — | — | 14.5 |
| LaSrCoO3 | 53.15 | 15.87 | 18.09 | — | 2.13 | — | 10.77 |
| LaSrCoFeO3 | 9.72 | 6.86 | 46.05 | — | 2.53 | 4.69 | 30.15 |
| LaCoO3-S | 4.62 | 3.74 | 33.21 | 4.06 | — | — | 54.37 |
| LaSrCoO3-S | 4.47 | 3.96 | 32.81 | 4.00 | 0.91 | — | 53.86 |
| LaSrCoFeO3-S | 4.32 | 3.23 | 33.76 | 3.18 | 1.76 | 2.18 | 51.85 |
图 7(B)为La1-xSrxCo1-yFeyO3催化剂硫化前后O1s的XPS谱图,其结合能及表面吸附氧含量列于表 3中,研究表明,低结合能(~529.5 eV)处的O1s峰归属为晶格氧物种Oβ(O2-),高结合能(~532.0 eV)处归属于表面吸附氧物种Oα(O2-和O-)。Oα和Oβ通常存在于缺陷型氧化物中[32-33],且表面吸附氧相对晶格氧具有较好的活性和移动性,这对碳烟催化氧化至关重要。当LaCoO3掺杂Sr后形成较多表面吸附氧(58.30%),表明Sr掺杂有利于形成丰富的表面吸附氧和氧空位,有利于提高催化氧化碳烟活性,这也与图 1的活性结果一致。当Sr、Fe同时掺杂时其表面吸附氧含量(60.60%)增加,结合H2-TPR结果可知,这主要是因为金属掺杂导致的电荷转移使表面吸附氧含量增加。值得注意的是,当各催化剂硫化后,表面吸附氧的含量增加,这主要由于SO2与催化剂中金属离子及表面吸附氧反应形成硫酸盐物种导致[23]。当LaCoO3硫化后,会形成较多的硫酸盐物种(S元素的原子百分含量90.29%),表明LaCoO3硫化后对表面吸附氧有较大消耗;而同时掺杂Sr、Fe时表面吸附氧含量相对于新鲜催化剂变化较小,表明Fe掺杂对表面吸附氧有一定保护,这也与EDS-mapping和表 3结果一致。综上所述,催化剂中毒可能主要来源于SO2对催化剂中Co2+/Co3+及表面吸附氧的硫酸化。
下载:
导出CSV
| Sample | BE of Co2p3/2 / eV | BE of O1s / eV | nFe3+/nFe / % | nOα/(nOα+nOβ) / % | |
| Oβ | Oα | ||||
| LaCoO3 | 780.1 | 529.05 | 532.00 | — | 53.80 |
| LaSrCoO3 | 780.2 | 528.90 | 531.55 | — | 58.30 |
| LaSrCoFeO3 | 779.8 | 528.85 | 531.20 | 42.5 | 60.60 |
| LaCoO3-S | 781.8 | 528.70 | 531.75 | — | 90.29 |
| LaSrCoO3-S | 780.6 | 528.70 | 531.85 | — | 77.39 |
| LaSrCoFeO3-S | 779.6 | 528.75 | 531.45 | 14.5 | 70.96 |
图 7(C,D)为La1-xSrxCo1-yFeyO3催化剂硫化前后Co2p的XPS谱图。各催化剂均明显表现出2个Co2p肩峰,分别归属为Co2p1/2和Co2p3/2。LaCoO3催化剂Co2p3/2的结合能为780.10 eV,而同时掺杂Sr、Fe后,Co2p3/2向低结合能移动至779.85 eV,这是由于Fe掺杂改变了钙钛矿的电荷平衡,而为补偿钙钛矿氧化物使其电中性,催化剂会形成更多高价Fe4+和氧空位以平衡电荷,这有利于催化氧化碳烟反应。值得注意的是,LaCoO3和LaSrCoO3硫化后,Co2p3/2向高结合能偏移,表明SO2对催化剂Co2p价态环境有一定影响,结合H2-TPR可知,这是由于SO2对Co2+/Co3+及表面吸附氧的硫酸化,导致催化剂结合能偏移。进一步由表 3可知,LaSrCoFeO3-S(14.5%)相对新鲜的LaSrCoFeO3(42.5%)中的Fe3+含量下降65.8%,表明LaSrCoFeO3-S抗硫性可能由于Fe3+与表面吸附氧及SO2共同作用形成了Fe2(SO3)3、Fe2(SO4)3。另一方面,LaSrCoFeO3-S中Co2p3/2结合能也相对新鲜催化剂LaSrCoFeO3变化较小,表明Fe掺杂有利于对Co2p价态环境的保护,即同时掺杂Sr、Fe形成的Fe3+有助于减少SO2对活性位点表面吸附氧和Fe4+毒化。
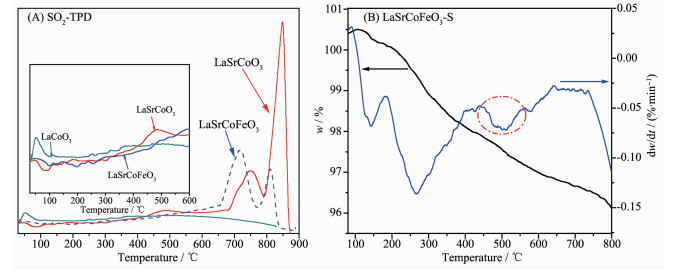
为进一步研究金属掺杂对吸脱附SO2的影响及硫酸盐的形式,对La1-xSrxCo1-yFeyO3催化剂的SO2-TPD曲线进行研究(图 8(A))。在50~200 ℃为SO2物理吸附态,250~500 ℃为中度硫酸盐物种分解,大于650 ℃为强粘结硫酸盐物种的分解[34-35]。LaCoO3催化剂中的SO2在整个温度范围内主要呈物理及中度吸附,即SO2与表面吸附氧形成弱或中度硫酸盐覆盖催化剂活性位点,导致催化剂失活,这也和XPS结果一致。当催化剂同时掺杂Sr、Fe时,在低温范围内对SO2吸附减少,有利于降低SO2对表面吸附氧的毒化,并在高温形成2个脱附峰,即670~800 ℃和800~860 ℃,这主要归属为SO2与Fe3+及表面吸附氧反应形成的强粘结硫酸盐Fe2(SO3)3和Fe2(SO4)3。图 8(B)为LaSrCoFeO3-S催化剂的热重-微商热重分析(TG-DTG)曲线,其在480 ℃处出现一个DTG失重峰,这主要归属为Fe2(SO3)3和Fe2(SO4)3的分解[36],表明LaSrCoFeO3催化剂抗硫性主要来源于形成的Fe2(SO3)3和Fe2(SO4)3保护了活性位点的表面吸附氧和Fe4+。
本研究利用柠檬酸-EDTA配位法制备了一系列钙钛矿催化剂La1-xSrxCo1-yFeyO3,展现出了较好的催化氧化碳烟活性。Sr掺杂LaCoO3有利于形成更多的表面吸附氧(O2-、O-)和氧空位,改善了低温氧化还原性能,提高了低温催化氧化碳烟活性,其Ti和Tm仅为284和347 ℃。掺杂Sr、Fe后低温氧化还原性能进一步提高,并形成更多的Fe4+离子,这有利于改善催化氧化碳烟活性。催化剂SO2中毒主要由于Co2+/Co3+和表面吸附氧的硫酸化(SO32-、SO42-);同时掺杂Sr、Fe后两者之间形成的相互作用能有效抑制硫酸盐在催化剂表面的沉积,减少SO2对催化剂的毒化。通过XPS和SO2-TPD发现,催化剂抗硫性主要来源于金属离子Fe3+与SO2结合形成硫酸盐(Fe2(SO3)3和Fe2(SO4)3),使SO2减少对活性组分表面吸附氧和Fe4+的毒化。通过TPO发现硫化后的LaSrCoFeO3仍具有较好的催化氧化碳烟活性,其Ti和Tm分别为320和361 ℃,表明Sr、Fe同时掺杂具有较好的低温催化氧化碳烟活性和良好的抗硫性能。
Li Z Q, Meng M, Zha Y Q, et al. Appl. Catal. B, 2012, 121:65-74
王世丹, 朱艺, 张海龙, 等.无机化学学报, 2014, 30(8):1827-1833 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20140813WANG Shi-Dan, ZHU Yi, ZHANG Hai-Long, et al. Chinese J. Inorg. Chem., 2014, 30(8):1827-1833 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20140813
饶成, 刘瑞, 冯小辉, 等.催化学报, 2018, 39(10):1683-1694RAO Cheng, LIU Rui, FENG Xiao-Hui, et al. Chin. J. Catal., 2018, 39(10):1683-1694
Van Setten B A A L, Makkee M, Moulijn J A. Catal. Rev. Sci. Eng., 2001, 43(8):489-564
牟一蒙, 梁红, 李树华, 等.无机化学学报, 2016, 32:602-608 doi: 10.11862/CJIC.2016.074MOU Yi-Meng, LIANG Hong, LI Shu-Hua, et al. Chinese J. Inorg. Chem., 2016, 32:602-608 doi: 10.11862/CJIC.2016.074
Matarrese R, Castoldi L, Lietti L, et al. Top. Catal., 2009, 52:2041-2046 doi: 10.1007/s11244-009-9400-4
Shangguan W, Teraoka Y, Kagawa S. Appl. Catal. B, 1998, 16:149-154 doi: 10.1016/S0926-3373(97)00068-4
Teraoka Y, Nakano K, Shangguan W, et al. Catal. Today, 1996, 27:107-113 doi: 10.1016/0920-5861(95)00177-8
Fang S Q, Wang L, Sun Z C, et al. Catal. Commun., 2014, 49:15-19 doi: 10.1016/j.catcom.2014.01.029
Li S X, Kato R, Wang Q, et al. Appl. Catal. B, 2010, 93:383-386 doi: 10.1016/j.apcatb.2009.10.012
Yi Y N, Liu H, Chu B X. et al. Chem. Eng. J., 2019, 369:511-521 doi: 10.1016/j.cej.2019.03.066
Ji K M, Dai H X, Deng J G, et al. Chem. Eng. J., 2013, 214:262-271 doi: 10.1016/j.cej.2012.10.083
Liu Y A, Zheng H T, Liu J R, et al. Chem. Eng. J., 2002, 89:213-221 doi: 10.1016/S1385-8947(02)00013-X
高永华, 高利珍, 崔佳丽, 等.太原理工大学学报, 2017, 48:747-752GAO Yong-Hua, GAO Li-Zhen, CUI Jia-Li, et al. Journal of Taiyuan University of Technology, 2017, 48:747-752
Liu F D, He H, Zhang C B. Chem. Commun., 2008, 164:2043-2045
Fabrizioli P, Burgi T, Baiker A. J. Catal., 2002, 206:143-154 doi: 10.1006/jcat.2001.3475
Liu F D, Shan W P, Lian Z H, et al. Appl. Catal. B, 2018, 230:165-176 doi: 10.1016/j.apcatb.2018.02.052
Li Z Q, Meng M, Li Q A, et al. Chem. Eng. J., 2010, 164:98-105 doi: 10.1016/j.cej.2010.08.036
Jin R B, Liu Y, Wang Y, et al. Appl. Catal. B, 2014, 148-149:582-588 doi: 10.1016/j.apcatb.2013.09.016
Yu C L, Huang B C, Dong L F, et al. Chem. Eng. J., 2017, 316:1059-1068 doi: 10.1016/j.cej.2017.02.024
Li S H, Huang B C, Yu C L, et al. Catal. Commun., 2017, 98:47-51 doi: 10.1016/j.catcom.2017.04.046
Liu H, Fan Z X, Sun C Z, et al. Appl. Catal. B, 2019, 244:671-683 doi: 10.1016/j.apcatb.2018.12.001
Jiang L J, Liu Q C, Ran C J, et al. Chem. Eng. J., 2019, 370:810-821 doi: 10.1016/j.cej.2019.03.225
Yu X H, Wang L Y, Chen M Z, et al. Appl. Catal. B, 2019, 254:246-259 doi: 10.1016/j.apcatb.2019.04.097
Liang H, Mou Y M, Zhang H W, et al. Catal. Today, 2017, 281:477-481 doi: 10.1016/j.cattod.2016.05.015
Li K B, Li X J, Zhu K G, et al. J. Appl. Phys., 1997, 81:6943-6947 doi: 10.1063/1.365234
Yamaguchi T, Jin T, Tanabe K. J. Phys. Chem., 1986, 90:3148-3152 doi: 10.1021/j100405a022
Kostoglou M, Housiada P, Konstandopoulos A G. Chem. Eng. Sci., 2003, 58:3273-3283 doi: 10.1016/S0009-2509(03)00178-7
Liang H, Hong Y X, Zhu C Q, et al. Catal. Today, 2013, 201:98-102 doi: 10.1016/j.cattod.2012.04.036
Li W, Zhang C, Li X, et al. Chin. J. Catal., 2018, 39(10):1653-1663 doi: 10.1016/S1872-2067(18)63099-2
Li B, Ren Z Y, Ma Z X, et al. Catal. Sci. Technol., 2015, 6:1719-1725
Tan R Q, Zhu Y F. Appl. Catal. B, 2005, 58:61-68 doi: 10.1016/j.apcatb.2004.12.003
Zhao Z, Yang X, Wu Y. Appl. Catal. B, 1996, 8:281-298 doi: 10.1016/0926-3373(95)00067-4
Chen L Q, Li R, Li Z B, et al. Catal. Sci. Technol., 2017, 7:3243-3257 doi: 10.1039/C7CY00672A
Peng Y, Wang D, Li B, et al. Environ. Sci. Technol., 2017, 51:11943-11949 doi: 10.1021/acs.est.7b03309
Machado L C, Marins A A L, Muri E J B, et al. J. Therm. Anal. Calorim., 2009, 97(1):289-296

图 3 La1-xSrxCo1-yFeyO3催化剂的FT-IR谱图
Figure 3 FT-IR spectra of La1-xSrxCo1-yFeyO3 catalysts

图 5 La1-xSrxCo1-yFeyO3催化剂硫化后的EDS-mapping照片
Figure 5 EDS-mapping photographs of La1-xSrxCo1-yFeyO3 catalysts after vulcanization

图 6 催化剂La1-xSrxCo1-yFeyO3的H2-TPR谱图
Figure 6 H2-TPR profiles of La1-xSrxCo1-yFeyO3 catalysts

图 8 La1-xSrxCo1-yFeyO3催化剂的(A) SO2-TPD和(B) TG-DTG
Figure 8 (A) SO2-TPD and (B) TG-DTG profiles of La1-xSrxCo1-yFeyO3 catalysts
表 1 催化剂硫化前后催化氧化碳烟活性对比
Table 1. Comparison of catalytic oxidation soot activity before and after catalyst vulcanization
| Sample | Ti / ℃ | Tm / ℃ | Tf / ℃ |
| LaCoO3 | 312 | 387 | 426 |
| LaSrCoO3 | 284 | 347 | 404 |
| LaCoFeO3 | 323 | 407 | 475 |
| LaSrCoFeO3 | 307 | 368 | 421 |
| LaCoO3-S | 421 | 482 | 500 |
| LaSrCoO3-S | 321 | 381 | 461 |
| LaSrCoFeO3-S | 320 | 361 | 460 |
| Soot | 400 | 586 | 675 |
 下载: 导出CSV
下载: 导出CSV
表 2 La1-xSrxCo1-yFeyO3催化剂硫化前后各元素相对原子百分含量
Table 2.
Relative atomic contents of elements for catalyst La1-xSrxCo1-yFeyO3 after vulcanization
| Sample | La | Co | O | S | Sr | Fe | C |
| LaCoO3 | 53.21 | 12.23 | 18.81 | — | — | — | 14.5 |
| LaSrCoO3 | 53.15 | 15.87 | 18.09 | — | 2.13 | — | 10.77 |
| LaSrCoFeO3 | 9.72 | 6.86 | 46.05 | — | 2.53 | 4.69 | 30.15 |
| LaCoO3-S | 4.62 | 3.74 | 33.21 | 4.06 | — | — | 54.37 |
| LaSrCoO3-S | 4.47 | 3.96 | 32.81 | 4.00 | 0.91 | — | 53.86 |
| LaSrCoFeO3-S | 4.32 | 3.23 | 33.76 | 3.18 | 1.76 | 2.18 | 51.85 |
下载: 导出CSV
表 3 La1-xSrxCo1-yFeyO3催化剂硫化前后Co2p3/2, O1s结合能和Fe3+、Oα百分含量
Table 3. Binding energies (BE) of Co2p3/2 and O1s, and the content of Fe3+ and Oα for the La1-xSrxCo1-yFeyO3 catalysts
| Sample | BE of Co2p3/2 / eV | BE of O1s / eV | nFe3+/nFe / % | nOα/(nOα+nOβ) / % | |
| Oβ | Oα | ||||
| LaCoO3 | 780.1 | 529.05 | 532.00 | — | 53.80 |
| LaSrCoO3 | 780.2 | 528.90 | 531.55 | — | 58.30 |
| LaSrCoFeO3 | 779.8 | 528.85 | 531.20 | 42.5 | 60.60 |
| LaCoO3-S | 781.8 | 528.70 | 531.75 | — | 90.29 |
| LaSrCoO3-S | 780.6 | 528.70 | 531.85 | — | 77.39 |
| LaSrCoFeO3-S | 779.6 | 528.75 | 531.45 | 14.5 | 70.96 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




