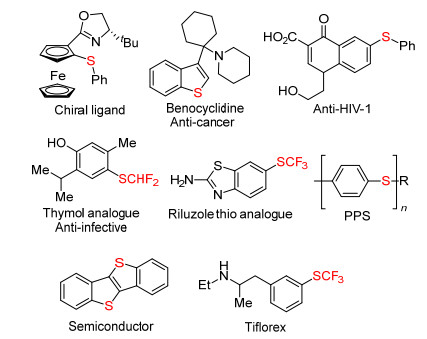
Figure 1.
Applications of aryl and vinyl sulfides

Aryl and vinyl sulfides are prevalent in natural products or bioactive molecules, [1] and also have wide application in a range of fields such as drug development, [2] organic material, [3] ligand design[4] or polymer science[5] (Figure 1). As a consequence, the development of efficient synthetic methods for the construction of aryl and vinyl sulfides has gained significant interest among medicinal and synthetic organic chemists over the past few decades.[6] Traditionally, transition-metal-catalyzed cross-coupling between C— H/C—H and various sulfur sources such as thiols, sulfonyl chlorides, disulfides, sulfonyl hydrazides, and other sulfur-containing reagents is well known.[7] In spite of some great advantages, some of these processes could suffer from certain drawbacks, including pre-functionalization of reactants, harsh reaction conditions, and toxic metal catalysts.
From the point of "atom economy" and "green chemistry", the direct transition metal-free sulfenylation of C—H bond can avoid some defects of transition-metal-catalyzed reactions. This protocol has attracted much attention in recent years and many significant research achievements have been presented, [8] which contains several reviews on the transition metal-free sulfenylation of various substrates including heteroarenes, indoles, pyrroles with sundry sulfur reagents from various views.[9] The recent five-year progress in direct sulfenylation of C—H bond on alkenes and arenes under transition metal-free conditions is reviewed and the corresponding reaction mechanisms in detail is discussed.
Sulfur-containing aromatic compounds are attracting much attention from medical and organic chemists because of their prevalence in natural products, pharmacologically active compounds and organic materials as ubiquitous structural motifs.[10] Therefore, various accesses to construction of C—S bond have been developed to synthesize aryl sulfides, and direct C—H sulfenylation under transition metal-free conditions is more practical and economical among these approaches. At present, different sulfenylating regents such as thiols, disulfides, thioethers, sulfonyl hydrazides, sulfonyl chlorides, sodium sulfinates, etc. have been employed for sulfenylation of arenes.
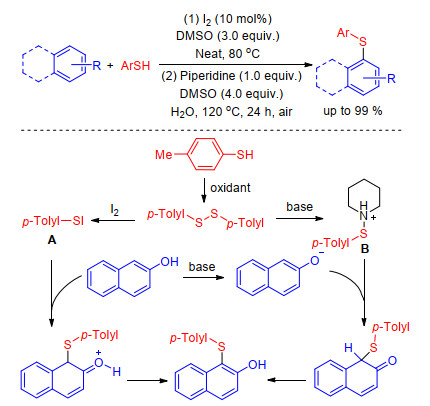
As the easily available and high atom economy sulfur sources, thiols and thiones are often applied for the sulfenylation of a range of substrate such as alkanes, [11] indoles, [12] imidazoheterocycles, [13] etc. Iodine-catalyzed sulfenylation of electron-rich arenes with various thiophenols in the presence of dimethyl sulfoxide (DMSO) as the oxidant under solvent-free conditions is described by Peddinti[14] and co-workers in 2015. Subsequently, Wang[15] and Xiao[16] groups discovered that this transformation can go smoothly in the presence of inexpensive, environment friendly NaI or piperidine using air as oxidant. Sensitive and active free hydroxyl and amino as well as other functional groups were well tolerated under these reaction conditions.
The mechanisms of aforementioned reaction that authors proposed in articles are very similar (Scheme 1). Initially the aryl thiols were converted to disulfides under oxidizing conditions. Immediately the formed disulfide reacts with iodine or base to form the electrophilic intermediates A and B which is attacked by electron-rich arenes forming desired compounds. Though other possible reaction pathway can't be ruled out, this mechanism for sulfenylation of electron-rich arenes with thiophenols was widely accepted.
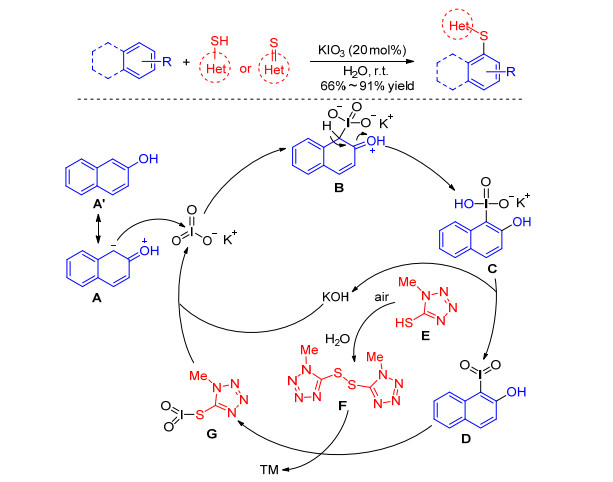
Unlike thiophenol, most of the heterocyclic thiols and thiones are odorless, stable compounds and are also useful precursors for synthesizing a lot of biologically active compounds.[17] In spite of so many merits, sulfenylation of arenes with heterocyclic thiols and thiones is still a challenging task and there are only few reports. In 2019, Lin group[18] disclosed the KIO3-catalyzed direct sulfenylation of phenol and arylamine derivatives with heterocyclic thiols and thiones in water at room temperature. The mechanism proposed by authors is described in Scheme 2. Firstly, the resonance of 2-naphthol A couples KIO3 to give the intermediate C through compound B. Further formation of compound D occurs by releasing KOH, and the intermediate D reacts with the disulfide F generated in situ affording the target products and compound G. Finally, the reaction of KOH and intermediate G allowed the regeneration of the catalyst (Scheme 2).
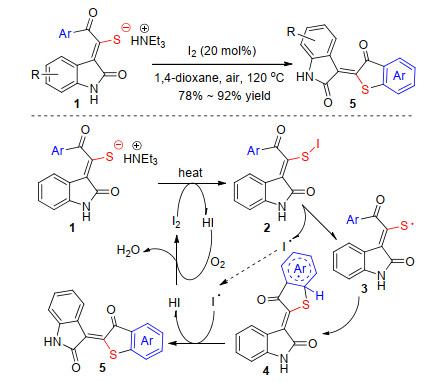
Despite the success of C—H bond intermolecular sulfenylated reactions, transition metal-free catalyzed intramolecular sulfenylation of C—H bond still provides a great challenge in modern organic synthesis. A facile method for the synthesis of benzo[b]thiophenes via intramolecular sulfenylation has been developed by Li and co-workers (Scheme 3).[19] Mechanism research indicates that substrate 1 undergoes S—I bond formation to generate intermediate 2, followed by homolysis of the S—I bond to yield the radical intermediate 3. The intramolecular radical coupling/cyclization between S-radical and arenes occurred through a single-electron transfer process to access the final product 5 via deprotonation. Under the optimized reaction conditions, electron-deficient arenes also can be sulfenylated smoothly in good to excellent yields.
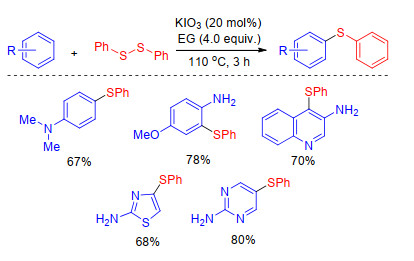
As the intermediate of many sulfenylation mechanisms with thiols, disulfides could be directly used as sulfur source to sulfenylate arenes and other compounds.[20] Braga and co-workers[21] outlined a new KIO3-catalyzed eco- friendly methodology for the sulfenylation of arenes (Scheme 4). This benign and robust protocol is performed with a non-toxic and easily handled catalyst with high atom-economy.
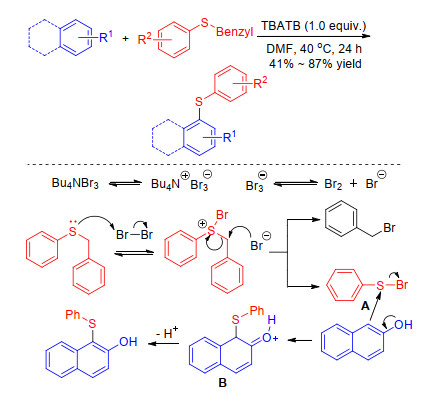
Barman and Yorimitsu group separately disclosed an efficient sulfenylation of arenes with alkyl aryl sulfides in the presence of feat promoters. The reaction substrate scope and mechanism are all different. Barman group[22] found that tetrabutylammonium tribromide (TBATB) can mediate the coupling reaction of arylbenzylsulfides with electron-rich arenes. The possible mechanism is that arylbenzylsulfide firstly reacts with Br2 generated in situ to form the electrophilic species RS-Br. The reactive sulfenyl bromide can react with arenes to form intermediate B, which further deprotonation forms the desired product (Scheme 5).
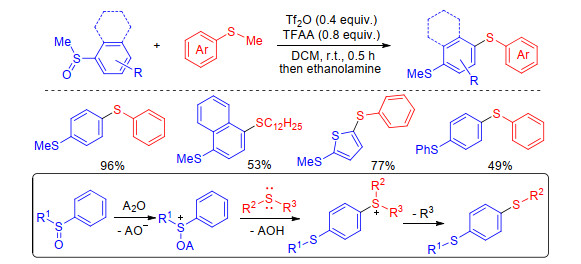
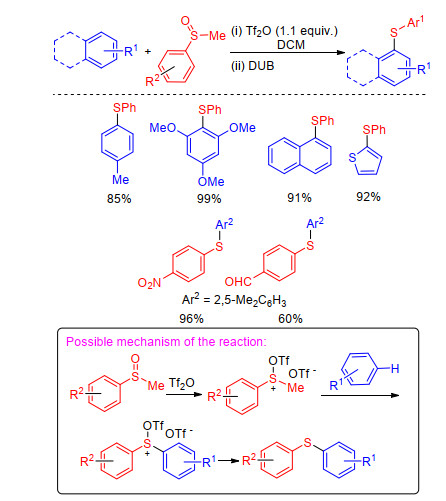
However, Yorimitsu group[23] discovered that the combination of Tf2O and trifluoroacetic anhydride (TFAA) could promote C—H sulfenylation of aryl sulfoxides with alkyl aryl sulfides. The reaction began with Pummerer-type activation of aryl sulfoxides followed by nucleophilic attack of alkyl aryl sulfides. The formed sulfonium salts were demethylated by ethanolamine to yield the desired products (Scheme 6). The salient features of the present protocol are simplicity, high efficiency, excellent regioselective and is compatible with various (hetero)arenes.
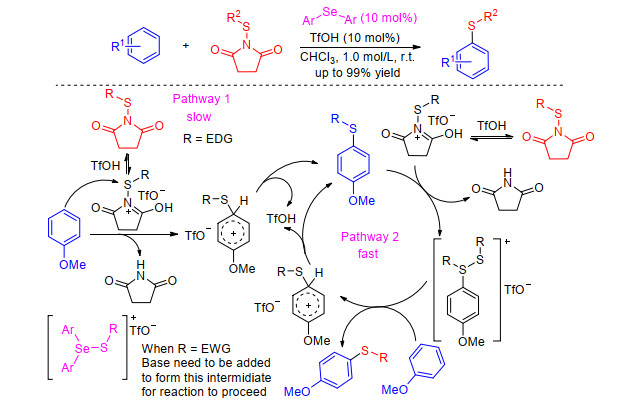
In 2015, Cossy and co-workers[24] have demonstrated that trifluoroacetic acid (TFA) can promote C—H sulfenylation of electron-rich arenes using N-(thio)succin- imides. Although it can produce various diarylsulfides and alkyl aryl sulfides, this method employed superstoichiometric quantities of trifluoroacetic acid to activate N-(thio)succinimides. While Gustafson group[25] modified the protocol and reported the combination of selenide ether and 10 mol% TfOH can result in rapid and robust conversions across several different arene classes (Scheme 7). They demonstrated that the incorporation of electron-rich sulfenyl groups proceeded in the absence of a Lewis base, with kinetic studies indicating an autocatalytic mechanism. The incorporation of electron-poor sulfenyl groups demonstrated little autocatalysis necessitating the use of Lewis base.
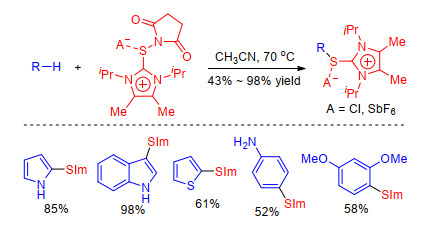
Recently, Alcarazo et al.[26] achieved C—H sulfenylation of electron-rich (hetero)arenes with newly prepared succinylthioimidazolium salts and synthesized a series of arylthioimidazolium salts which can react with Grignards affording unsymmetrical diaryl sulfides (Scheme 8). The synthetic protocol is modular, scalable and highly yielding, and provides access to sulfides that are not easy to obtain through the existing methodologies.
Gustafson's work can also be applied for the trifluoromethylthiolation of arenes replacing N-(thio)succinimide with N-trifluoromethylthiosaccharin as sulfenylating reagents.[25] With good lipophilicity and good bioavailability SCF3 group has been considered as one of privileged fragments that can improve the pharmacokinetic and physicochemical properties of drug molecules.[27] Consequently, great effort has been intensively devoted to exploring efficient methods for the incorporation of SCF3 group into molecules.[28] Among them, direct C—H trifluoromethylthiolation represents one of the most straightforward and promising approaches using trifluoromethylthiolating reagents, including CF3SCl, ArNHSCF3, CF3SO2Na, N-trif- luoromethylthioimide, (PhSO2)NSCF3, N-trifluoromethyl- thiosaccharin etc.
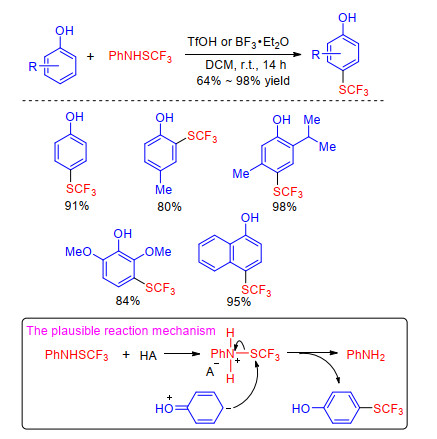
In 2015, Jereb group[29] reported that the trifluoromethylthiolation of variously substituted phenols was accomplished by PhNHSCF3 in the presence of TfOH or BF3• Et2O. The trifluoromethylthiolation was exclusively para- selective. The para-substituted phenols gave the ortho- substituted SCF3-products, while multiple substituted phenols afforded the corresponding products according to the Mills-Nixon effect. Authors speculated that the reaction selectivity indicates an electrophilic pathway (Scheme 9). In addition, Iskra and co-workers[30] discovered that p- chloro-substituted PhNHSCF3 was more stable and effective because it could not react by trifluoromethylthiolation of itself.
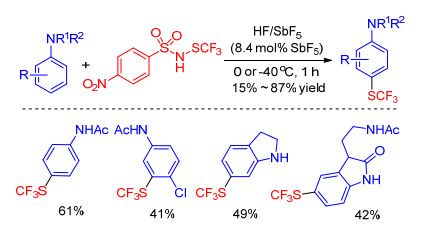
Thibaudeau and co-workers[31] discovered that incorporation electron withdrawing group like sulfonyl, carbonyl between nitrogen and sulfur atom could raise its reactivity when they explored the trifluoromethylthiolation of aromatic amines with ArNHSCF3. Therefore, a novel trifluoromethylthiolating reagent NSCF3 sulfenamide was synthesized and applied for the trifluoromethylthiolation of arylamine with a broad substrate scope such as alkaloida or steroids under superacid conditions (Scheme 10). Authors don't present a specific reaction process, but NMR spectroscopy studies revealed the involvement of dicationic superelectrophilic intermediates.
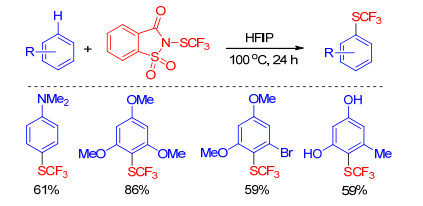
Recently, Xu group[32] developed an efficient hydrogen bonding assisted regioselective trifluoromethylthiolation of electron-rich arenes using N-trifluoromethylthiosaccharin. The hydrogen-bonding-network generated between hexafluoroisopropanol (HFIP) and trifluoromethylthiolating reagent could enhance the electrophilicity of the SCF3 electrophile, so the trifluoromethylthiolation underwent well under metal-free and catalyst-free conditions and showed good functional group tolerance (Scheme 11). Then, Shen's group[33] discovered that 2, 2, 2-trifluoro- ethanol (TFE) could also promote the reaction at lower temperature in higher yields.
In 2016, Shen and co-workers[34] reported a shelf-stable, easily prepared trifluoromethylthiolating reagent N-tri- fluoromethylthio-dibenzenesulfonimide exhibiting higher reactivity than other known electrophilic trifluoromethylthiolating reagents. In the absence of any additive, this reagent can react with a wide range of arenes including these common trifluoromethylthiolate unactivated and styrene under mild conditions (Scheme 12). Moreover, the reaction of some electron-rich arenes like naphthol derivatives with N-trifluoromethylthio-dibenzenesulfonimide of- fered the corresponding dearomative trifluoromethylthiolated product instead of the direct C—H trifluoromethylthiolating product.
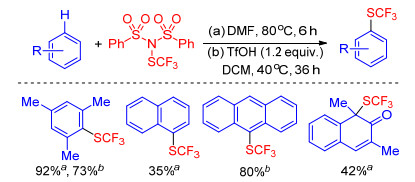
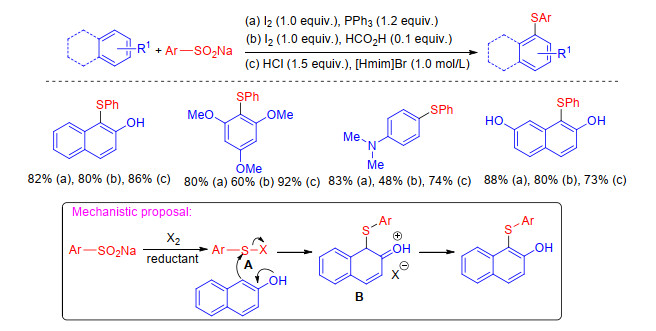
In the specified reaction conditions, sodium sulfinates can be reduced to disulfides which are classical sulfenylating agents. Therefore, sodium sulfinates as desirable sulfur source have been extensively used to sulfenylate C—H bond because they are readily accessible and environment- friendly. In 2015, Lin and co-workers[35] described the synthesis of diaryl sulfide derivatives via the I2-PPh3 system mediated sulfenylation of substituted naphthols/phe- nylamines in water under air condition. In this program, using a range of arylsulfinic acid salts as sulfur source, structurally diversed naphthol/aniline thioethers can be smoothly produced in moderate to good yields. Xiao group[36] found that the sulfenylation underwent well replacing triphenylphosphine with formic acid as reductant, while Lu group[37] applied [Hmim]Br as both solvent and reducer avoiding the stoichiometric toxic reductant and iodine. The reaction mechanism proposed by these groups is strikingly consistent. Initially, sodium sulfinate was reduced to disulfides in the presence of reductant which reacts with X2 to form electrophilic species ArSX A. The species ArSX is attacked by substrate to yield the intermediate B, which releases a proton to afford the desired product (Scheme 13).
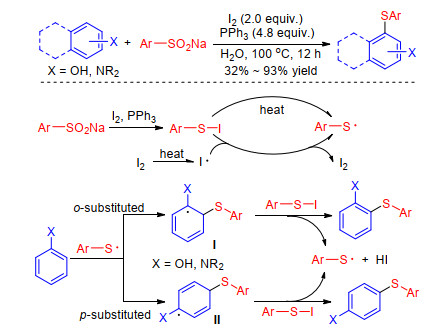
Yi group[38] also disclosed the synthesis of diaryl sulfides using odorless sodium sulfinates as the sulfur source in an aqueous I2-PPh3 system, but the authors deduced that the reaction involved a free radical process on the basis of control experiments and density functional theory (DFT) calculations. Although the exact mechanism remained to be elucidated, a possible pathway was proposed in Scheme 14. At first, arylsulfenyl radicals are formed via the homolysis of I2 or ArSI in situ generated in the I2-PPh3 system. Then, an arylsulfenyl radical adds to the arene to produce intermidate Ⅰ or Ⅱ. Finally, the target products are formed by the reaction of ArSI and intermidate Ⅰ or Ⅱ following the formation of arylsulfenyl radicals and HI (Scheme 14).
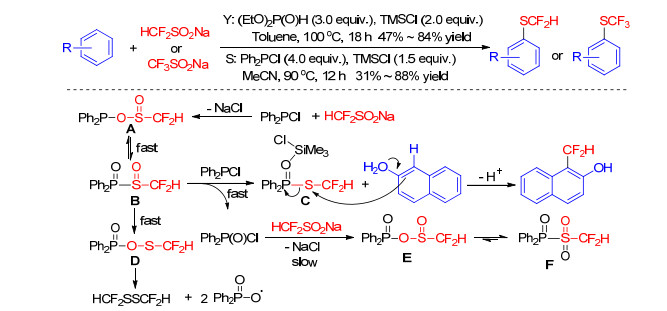
Furthermore, Yi group[39] also introduced a straightforward difluoromethylthiolation and trifluoromethylthiolation of arenes with sodium difluoromethanesulfinate or sodium trifluoromethanesulfinate in the presence of (EtO)2P(O)H and TMSCl. On the base of NMR spectroscopy, authors deduced that fluoroalkylsulfinate salts were reduced by (EtO)2P(O)H and TMSCl to form HCF2S+or CF3S+ for fluoromethylthiolation of aromatics, but didn't elaborate the specific process.
Before that, Shibata and co-workers[40] reported the difluoromethylthiolation of phenols and naphthols under the Ph2PCl-TMSCl system. They hypothesized that the reaction of HCF2SO2Na with Ph2PCl firstly generated intermediate A, which converted to intermediate B via an intramolecular rearrangement. The reactive species C formed by reduction of B can be highly activated in the presence of TMSCl as Lewis acid for electrophilic difluoromethylthiolation (Scheme 15). Subsequently, Zhao et al.[41] demonstrated that this process worked well with PCl3 or PhPCl2 in the absence of TMSCl and Shibata et al.[42] discovered that trifluoromethanesulfonyl chloride was also an efficient SCF3 source in the presence of phosphine reagent, but all only for electron-rich aromatics excluding arenes.
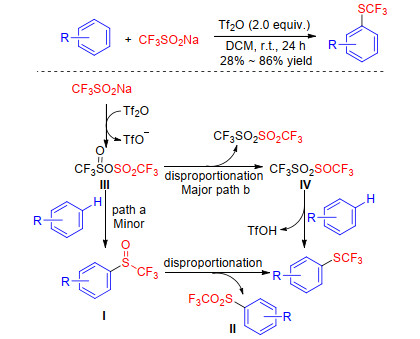
In 2018, Yi group[43] developed another method for the direct trifluoromethylthiolation using CF3SO2Na. A close examination of the literature and controlled experiment results showed that CF3SO2Na converted to the intermediate Ⅲ in the presence of triflic anhydride, which could offer the final product in two potential paths. In path a, trifluoromethyl sulfoxide Ⅰ partly contributed to the formation of SCF3 product via disproportionation under the anhydride system. Therefore, the intermediate Ⅲ also underwent self-disproportionation to form the reactive species Ⅳ in path b. Finally, an electrophilic trifluoromethylthiolation reaction of Ⅳ with arenes mainly contributed to the formation of desired product (Scheme 16). This catalytic system has been successfully employed to the direct trifluoromethylthiolation of a wide range of arenes including electron-deficient ones.
Dimethyl sulfoxide is often employed for sulfenylation reactions, but other sulfoxides are rarely used as sulfur sources. In 2016, Procter and co-workers[44] achieved the sulfenylation of arenes using bench-stable, convenient sulfoxides as sulfur source (Scheme 17). The readily available sulfoxides is once activated to form sulfoxonium salts that serve as excellent sulfur electrophiles for coupling with a variety of aryl partners including arenens, heteroarenens and alkenes. The method allows the incorporation of a variety of functional groups in coupling partner, is operationally simple and suitable for the construction of complex diarylsulfides.
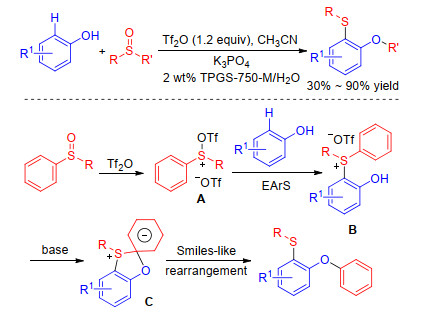
Additionally, Huang group[45] developed an environmentally benign method for the sulfenylation of phenols in 2017. They also used sulfoxides as sulfur source, but the process was mediated by electrophilic aromatic substitution and subsequent Smiles-like rearrangement different from Procter's work. Authors hypothesized that sulfoxides was also activated by trifluoromethanesulfonic anhydride to form sulfoxonium salts A which was attacked by phenols at sulfur giving rise to the intermediate B. Then such intermediates went a Smiles-like rearrangement affording versatile products (Scheme 18). This method applies to not only diaryl sulfoxides but also alkyl aryl sulfoxides affording the phenoxyaryl sulfides, which could be converted to many important ligands under aqueous micellar conditions.
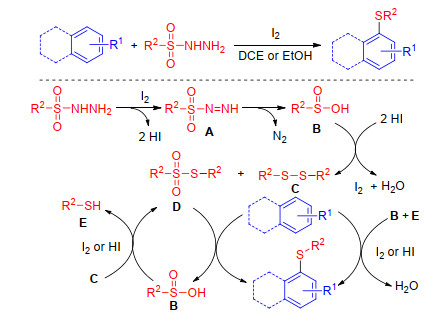
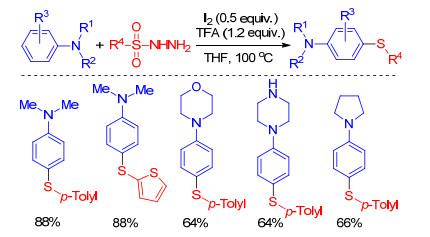
Sulfonyl hydrazides are useful reagents for C—S bond formation in organic synthesis.[46] Generally speaking, electrophilic thiodiazonium (RSN2I) or sulfenyl iodide (RSI) reagents which can be generated through the reactions between I2 and sulfonyl hydrazides can react with carbon nucleophilic reagents to form various C—S bonds. Yan group[47] outlined an iodine/TFA-mediated synthesis of aromatic thioethers from arylamine and sulfonyl hydrazides via C—H sulfenylation (Scheme 19). In this procedure, various substituents on the sulfonyl hydrazides and arylamines are tolerated in the sulfenylation, but not including 1-phenyl-1H-pyrrole, N-methylaniline and aniline.
In 2016, Xiao and co-workers[48, 49] consecutively published two articles on the sulfenylation of arenes using sulfonyl hydrazides. Electron-rich arenes and flavonoid derivatives were converted to desired products in moderate to good yields. A nearly identical mechanism was proposed. Initially, sulfonyl hydrazide decomposes into disulfide C and thiosulfonate D in the presence of iodine. The key intermediate D reacts with electron-rich arenes to produce the products as well as sulfinic acid B when I2 or HI is added. Intermediate B then reacts with C to yield D and thiophenol E. The combination of B and E can be used as the sulfur source to convert substrates into the sulfenylated products in the presence of iodine (Scheme 20).
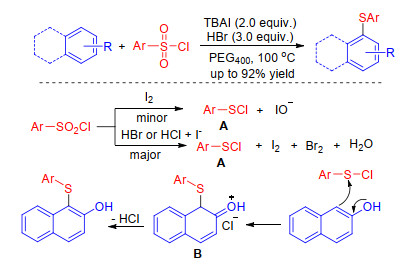
Lin group[50] found that sulfonyl chlorides were good sulfenylating reagents for the sulfenylation of electron-rich arenes in the presence of TABI/HBr. The desired products could be obtained in excellent yields by using widely available arylsulfonyl chlorides as the sulfur sources. A possible reaction mechanism is described in Scheme 21. HBr or HI which was formed by reaction between TBAI and acid, could reduce the arylsulfonyl chlorides to electrophilic ArSCl, which could react with arenes to give intermediates B. Then, the target product was formed by deprotonation. Alternatively, ArSCl could also be formed by reduction of arylsulfonyl chlorides with iodine, but this might be the minor pathway (Scheme 21).
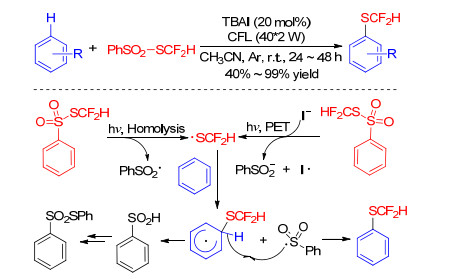
In 2018, Li group[51] described a visible light irradiated difluoromethylthiolation using the shelf-stable and readily available reagent PhSO2SCF2H. Inspired by this work in 2017, [52] authors speculated the homolytic fragmentation of PhSO2SCF2H with suitable light source and proposed a mechanism (Scheme 22). Under light irradiation, a difluoromethylthiyl radical resulted from either homolysis of PhSO2SCF2H or electron transfer event between PhSO2S- CF2H and iodide. Then the reaction of •SCF2H with arenes following the abstraction of hydrogen by PhSO2• affords the desired product. The phenylsulfinic acid was unstable and further transformed into the thiosulfonate. This protocol has successfully transformed a range of arenes into difluoromethylthioethers in the absence of transition metals and stoichiometric amount of additives.
Polysubstituted olefins are immanent structural units among biologically important drugs and natural products. Importantly, functionalized alkenes also contribute extensively to materials science and as building blocks in organic synthesis, in which the synthesis of sulfenyl olefins has attracted much attention because of its prevalent application in the synthesis of organic products in recent years.[53]
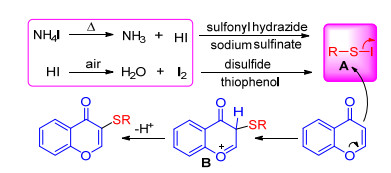
Flavone is a well-known natural product class in drug discovery with carbonyl-conjugated olefin function in its structure. Its electron-rich ring enables it to undergo well direct C—H sulfenylation. Until now, ammonium iodide- promoted sulfenylation of flavones with various sulfenylating reagents like sulfonyl hydrazide, thiophenol, sodium sulfinate, disulfide has been developed by Ge, [54, 57] Zhou, [55] Guo[56] et al. These reaction mechanisms were initiated from that NH4I was split into NH3 and HI at high temperature. The resulting HI was further oxidized by air to generate iodine, which then reacted with thiophenol to form electrophilic species RSI. Using sulfonyl hydrazide and sodium sulfinate as sulfur source, HI will promote the formation of electrophilic species RSI. Subsequently, RSI reacts further with electron-rich flavone to give reaction intermediate B, from which loss of a proton affords the final product (Scheme 23). Without doubt, flavone and derivatives can be sulfenylated by these sulfur reagents with external iodine.[58]
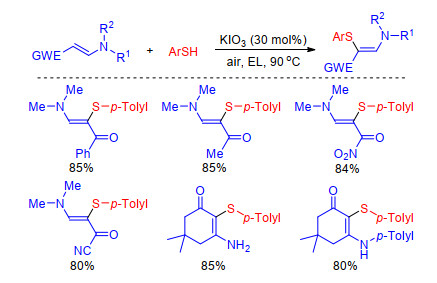
Moreover, Wan and co-workers[59] have realized the synthesis of polyfunctionalized aminothioalkenes via the direct C—H sulfenylation of enaminones in the presence of catalytic amount KIO3 in a green medium in 2016 (Scheme 24). A tentative mechanism authors proposed is similar to that Liu proposed for the KIO3-catalyzed sulfenylation of indoles.[60]
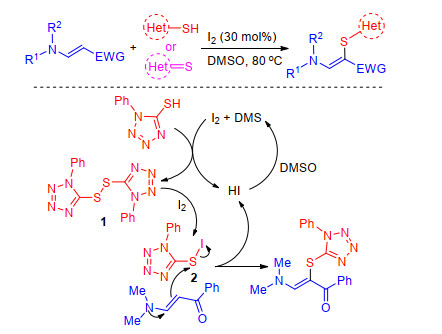
In 2017, Prabhu group[61] completed the sulfenylation of enaminones using iodine as catalyst and DMSO as oxidant. The presented method provides a straightforward approach to polyfunctionalized aminothioalkenes with a broad range of heterocyclic thiols and thiones. A tentative mechanism was proposed in Scheme 25. Heteroarylthiol reacts with iodine to form disulfide 1, which reacts with I2 to form an intermediate 2. In addition, nucleophilic displacement of an iodo group by enaminone forms product and byproduct HI, which is oxidized to iodine by DMSO. Based on the mechanism, Xiao and co-workers[62] achieved iodine- promoted three-component synthesis of substituted aminothioalkenes, in which enaminone is the intermediate of Michael-type addition of pyrrolidine to methyl propiolate and sulfenylated by disulfides.
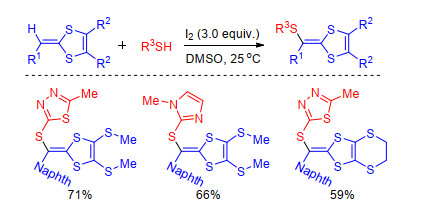
Recently, Zhu group[63] described the synthesis of novel thiadiazole-based DTF derivatives via iodine-mediated vinylic C(sp2)-H sulfenylation with heteroarylthiols throu- gh radical pathway. This new protocol is simple and effective for preparation DTF containing thiadiazole group using available material under mild conditions (Scheme 26).
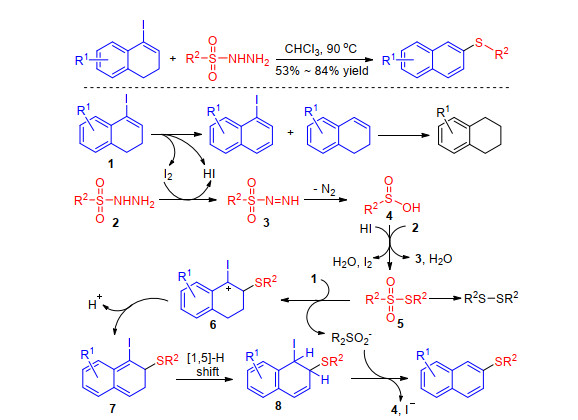
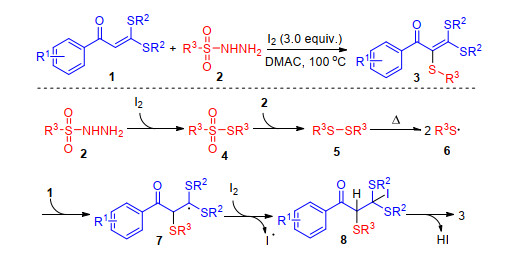
In 2016, Yang and co-workers[64] reported the by-produ- ct I2-catalyzed tandem redox-neutral sulfenylation/deio- dination/aromatization of cyclic iodoalkenes with sulfonyl hydrazides. In the absence of external catalysts and additives, a range of 4-iodo-1, 2-dihydronaphthalenes reacted with sulfonyl hydrazides to give structurally diverse 2-naphthyl thioethers in good yields. Mechanism study indicates that iodoalkene 1 decomposes upon heating to generate small amounts of aryl iodide, alkene, HI and I2. While the decomposition of sulfonyl hydrazide 2 to sulfinic acid 4 can take place upon heating, the iodine dramatically accelerates this process. Reduction of sulfinic acid 4 occurs either with sulfonyl hydrazide 2 or HI, and a small portion of the resulting thiosulfonate 5 is further reduced to give disulfide. Thiosulfonate 5 undergoes electrophilic addition to alkenyl iodide activated by iodine to generate carbocation 6, deprotonation of which gives dihydronaphthalene 7. Then [1, 5]-sigmatropic hydrogen shift occurs to generate alkene 8, from which elimination of HI gives the product (Scheme 27). The same group sequentially disclosed the sulfenylation of acyclic iodoalkene and bromoalkene which were the intermediates in deoxygenative iodization and olefination of ketones with sulfonyl hydrazides.[65]
In 2018, Liu and co-workers[66] achieved the synthesis of polythiolated alkenes via direct sulfenylation of the vinyl C—H bond employing sulfonyl hydrazines for the sulfenylating reagent. A mechanism is proposed in Scheme 28. First, the sulfonyl hydrazine can be transformed into disulfide 5 via the intermediate 4 resulting from a featured reduction in the presence of I2. The thermo-induced homolytic cleavage on 5 then provides the thiyl radical 6, which reacts with ketene dithioacetal leading to the formation of intermediate 7. The reaction of 7 and iodine provides intermediate 8, from which the successive elimination of HI yields products 3 (Scheme 28). The metals-free conditions, broad substrate tolerance, and simple operation ensure the present method a useful option for the synthesis of useful polythiolated alkene derivatives.
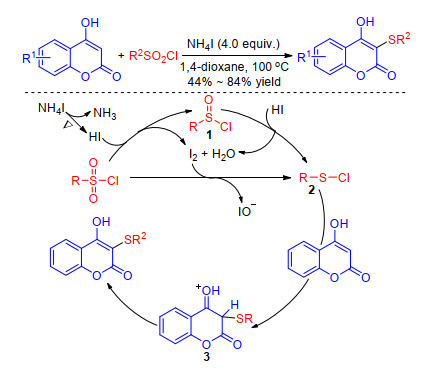
As the isomer of flavones, coumarins also have attracted immense attention as these compounds show a variety of biological and pharmacological activities. In 2017, with the same catalyst NH4I, Guo group[67] achieved the sulfenylation of 4-hydroxycoumarins using aryl- or alkyl-sulfonyl chlorides as the sulfur source. Similarly, NH4I firstly splits into NH3 and HI, and then HI reduces the sulfonyl chloride to the electrophilic species 2 which reacts with 4-hydroxy- coumarin to form intermediate 3. Finally, the product is obtained by deprotonation and aromatization of 3 (Scheme 29). In 2018, Zhou group[68] reported an efficient, transition metal-freed approach to the synthesis of sulfenylated coumarins using the same conditions as Wang's work[58] except for solvents. The iodine catalyzed sulfenylation is simple in operation, good functional group tolerance and use more environmentally water as solvent.
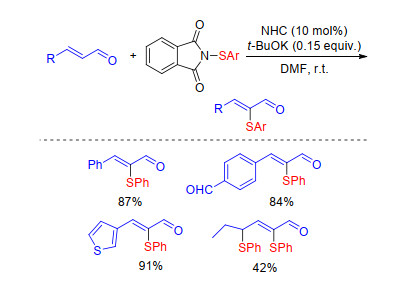
The sulfenylation reaction of acyclic α, β-unsaturated aldehydes with N-(arylthio)phthalimide has been developed by Chen and co-workers.[69] A wide variety of α-thioenals can be obtained with good yields and excellent Z-confi- guration using N-heterocyclic carbene (NHC) as catalyst under transition metal-free conditions (Scheme 30).
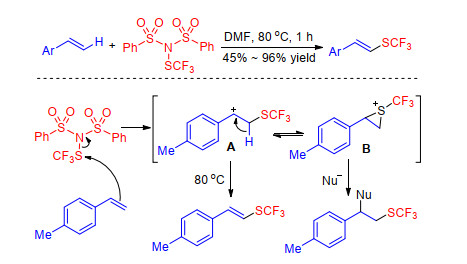
There are few research reports on the trifluoromethylthiolation reaction of olefins, most of which are one or more examples of substrate exploration in the research of trifluoromethylthiolation of aromatics.[40~42, 70] For example, Shen group[34] discovered that N-trifluoromethylthio- dibenzenesulfonimide can react with a wide range of styrenes in the absence of any additives. Initially, the trifluoromethylthiolating reagent is attacked by styrene to form benzylic cation intermediate A or sulfonium intermediate B. The intermediate A converts to target product by elimination of hydrogen, while intermediate B can be attacked by nucleophiles in reaction affording the difunctional product (Scheme 31).
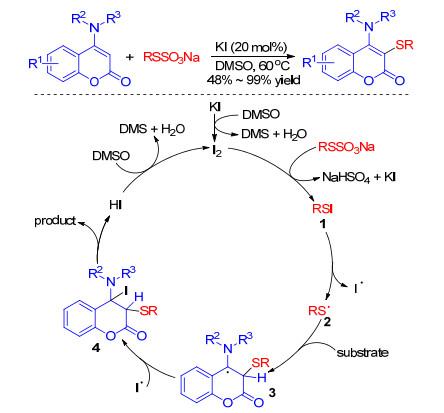
Moreover, an eco-friendly protocol for the KI-catalyzed regioselective sulfenylation of 4-anilinocoumarins with Bunte salts was established by Yang and co-workers.[71] A wide range of functional groups were tolerated well under the reaction conditions and the corresponding sulfenylated 4-anilinocoumarins were obtained in moderate to excellent yields (Scheme 32). Initially, the iodide ion was oxidized to iodine in the presence of DMSO. Then, the iodine reacted with Bunte salts to generate the electrophilic intermediate RSI which underwent a homolytic cleavage process to afford an iodine radical and a thiyl radical 2. The thiyl radical 2 reacted with 4-anilinocoumarins to give the carbon centered radical 3, which subsequently abstracted the iodine radical to give the intermediate 4. Then, the intermediate 4 loses a proton to give the desired products and HI. Finally, I was oxidized to I2 with DMSO to realize the catalytic cycle.
Herein we present an overview of the recent five-year progress in direct sulfenylation of C—H bonds on arenes and alkenes under transition metal-free conditions. Although various methods and sulfenylating reagents have been developed for the construction of aryl and vinyl sulfides, there are still lots of challenges that need to be addressed. Firstly, most of present methods are applied for the sulfenylations of arenes, especially the electron-rich arenes, and few researches on electron-poor arenes and olefins. Moreover, carbonyl-conjugated alkenes are the most commonly used substrates in the sulfenylation of alkenes, and the simple alkenes such as styrene are rarely used, especially its fluoroalkylthiolation. Furthermore, visible-light mediated photoredox catalysis and electrochemical system have been applied for the direct sulfenylation of alkanes and indoles, but there are not many applications in the sulfenylation and fluoroalkylthiolation of arenes and alkenes. Finally, the mechanisms of these reactions, which are reported so far, need to be further explored because specific reaction mechanisms could help us to design and achieve more efficient and eco-friendly sulfenylations. All of these need to be further explored and the results need to be further improved upon. The direct sulfenylation of arenes and alkenes is still a vigorous research field with both huge challenges and great prospect. We believe there will be more and more innovative achievements presented in the near future.
(a) Kvasnika, M.; Urban, M.; Dickinson, N. J.; Sarek, J. Nat. Prod. Rep. 2015, 32, 1303.
(b) Meng, D.; Chen, W.; Zhao, W. J. Nat. Prod. 2007, 70, 824.
(c) Cremlyn, R. J. An Introduction to Organosulfur Chemistry, Wiley, New York, 1996.
(d) Iino, H.; Usui, T.; Hanna, J.-I. Nat. Commun. 2015, 5, 6828.
(e) Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 11, 1596.
(a) Feng, M.; Tang, B.; Liang, S. H.; Jiang, X. Curr. Top. Med. Chem. 2016, 16, 1200.
(b) Kim, S.; Dahal, N.; Kesharwani, T. Tetrahedron Lett. 2013, 54, 4373.
(a) Boyd, D. A. Angew. Chem., Int. Ed. 2016, 55, 15486.
(b) Wu, D.; Pisula, W.; Haberecht, M. C.; Feng, X.; Müllen, K. Org. Lett. 2009, 11, 5686.
(c) Yang, S. M.; Shie, J. J.; Fang, J. M.; Nandy, S. K.; Chang, Y. Y. J. Org. Chem. 2002, 67, 5208.
(a) Carretero, J. C. Chem. Commun. 2011, 47, 2207.
(b) Pellisier, H. RSC Catalysis Series 2, Royal Society of Chemistry, Cambridge, 2009.
(a) Kausar, A.; Zulfiqar, S.; Sarwar, M. I. Pol. Rev. 2014, 54, 185.
(b) Rahate, A. S.; Nemade, K. R.; Waghuley, S. A. Rev. Chem. Eng. 2013, 29, 471.
(c) Spassky, N. Phosphorus Sulfur Silicon Relat. Elem. 1993, 74, 71.
(a) Hartwig, J. F. Nature 2008, 455, 314.
(b) Lu, Q.; Zhang, J.; Wei, F. L.; Qi, Y.; Wang, H.; Liu, Z.; Lei, A. Angew. Chem., Int. Ed. 2013, 52, 7156.
(c) Lu, Q.-Q.; Zhang, J.; Zhao, G.-L.; Qi, Y.; Wang, H.-M.; Lei, A. J. Am. Chem. Soc. 2013, 135, 11481.
(a) Beletskaya, I. P.; Ananikov, V. P. Eur. J. Org. Chem. 2007, 2007, 3431.
(b) Ley, S. V.; Thomas, A. W. Angew. Chem., Int. Ed. 2003, 42, 5400.
(c) Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T. S. A.; Liu, X. Chem. Soc. Rev. 2015, 44, 291.
(a) Zhang, S.-N.; Yang, S.-H.; Huang, L.-H.; Zhao, B.-L.; Cheng, K.; Qi, C.-Z. Chin. J. Org. Chem. 2015, 35, 2259(in Chinese).
(张诗浓, 杨胜虎, 黄乐浩, 赵保丽, 程凯, 齐陈泽, 有机化学, 2015, 35, 2259.)
(b) Xu, X.-M.; Chen, D.-M.; Wang, Z.-L. Chin. J. Org. Chem. 2019, 39, 3338(in Chinese).
(徐鑫明, 陈德茂, 王祖利, 有机化学, 2019, 39, 3338.)
(c) Dalpozzo, R. Org. Chem. Front. 2017, 4, 2063.
(d) Freckleton, M.; Baeza, A.; Benavent, L.; Chinchilla, R. Asian J. Org. Chem. 2018, 7, 1006.
(e) Sun, J.; Qiu, J.-K.; Zhu, Y.-L.; Guo, C.; Hao, W.-J.; Jiang, B.; Tu, S.- J. J. Org. Chem. 2015, 80, 8217.
(f) Sun, J.; Qiu, J.-K.; Jiang, B.; Hao, W.-J.; Guo, C.; Tu, S.-J. J. Org. Chem. 2016, 81, 3321.
(a) Liu, Y.-Y.; Xiong, J.; Wei, L. Chin. J. Org. Chem. 2017, 37, 1667(in Chinese).
(刘云云, 熊进, 韦丽, 有机化学, 2017, 37, 1667.)
(b) Dong, D.-Q.; Hao, S.-H.; Yang, D.-S.; Li, L.-X.; Wang, Z.-L. Eur. J. Org. Chem. 2017, 2017, 6576.
(c) Xu, X.-M.; Li, J.; Wang, Z.-L. Chin. J. Org. Chem. 2020, 40, 886(in Chinese).
(徐鑫明, 李家柱, 王祖利, 有机化学, 2020, 40, 886.)
(d) Jin, C.-A.; Xu, Q.; Feng, G.-F.; Jin, Y.; Zhang, L.-Y. Chin. J. Org. Chem. 2018, 38, 775(in Chinese).
(金城安, 徐庆, 冯高峰, 金阳, 张连阳, 有机化学, 2018, 38, 775.)
(e) Xu, X.-M.; Chen, D.-M.; Wang, Z.-L. Chin. Chem. Lett. 2020, 31, 49.
(a) Nakazawa, T.; Xu, J.; Nishikawa, T.; Oda, T.; Fujita, A.; Ukai, K.; Mangindaan, R. E. P.; Rotinsulu, H.; Kobayashi, H.; Namikoshi, M. J. Nat. Prod. 2007, 70, 439.
(b) Nielsen, S. F.; Olsen, G. M.; Liljefors, T.; Peters, D. J. Med. Chem. 2000, 43, 2217.
(c) Mori, T.; Nishimura, T.; Yamamoto, T.; Doi, I.; Miyazaki, E.; Osaka, I.; Takimiya, K. J. Am. Chem. Soc. 2013, 135, 13900.
(a) Varun, B. V.; Gadde, K.; Prabhu, K. R. Org. Lett. 2015, 17, 2944.
(b) Cao, H.; Yuan, J.; Liu, C.; Hu, X.-Q.; Lei, A.-W. RSC Adv. 2015, 5, 41493.
(c) Siddaraju, Y.; Prabhu, K. R. Org. Lett. 2016, 18, 6090.
(d) Siddaraju, Y.; Prabhu, K. R. J. Org. Chem. 2018, 83, 2986.
(e) Wang, D.; Liu, Z.; Wang, Z.; Ma, X.; Yu, P. Green Chem. 2019, 21, 157.
(f) Chen, Q.; Yu, G.; Wang, X.; Ou, Y.; Huo, Y. Green Chem. 2019, 21, 798.
(a) Ohkado, R.; Ishikawa, T.; Iida, H. Green Chem. 2018, 20, 984.
(b) Guo, W.; Tan, W.; Zhao, M.; Tao, K.; Zheng, L.-Y.; Wu, Y.; Chen, D.; Fan, X.-L. RSC Adv. 2017, 7, 37739.
(c) Zhang, H.; Bao, X.; Song, Y.; Qu, J.; Wang, B. Tetrahedron 2015, 71, 8885.
(d) Bai, F.-C.; Zhang, S.; Wei, L.; Liu, Y.-Y. Asian J. Org. Chem. 2018, 7, 371.
(a) Hiebel, M.; Berteina-Raboin, S. Green Chem. 2015, 17, 937.
(b) Siddaraju, Y.; Prabhu, K. R. J. Org. Chem. 2016, 81, 7838.
(c) Iida, H.; Demizu, R.; Ohkado, R. J. Org. Chem. 2018, 83, 12291.
(d) Yuan, Y.; Cao, Y.; Qiao, J.; Lin, Y.; Jiang, X.; Weng, Y.; Tang, S.; Lei, A. Chin. J. Chem. 2019, 37, 49.
(e) Rahaman, R.; Das, S.; Barman, P. Green Chem. 2018, 20, 141.
Parumala, S. K. R.; Peddinti, R. K. Green Chem. 2015, 17, 4068. doi: 10.1039/C5GC00403A
Wang, H.-H.; Shi, T.; Gao, W.-W.; Wang, Y.-Q.; Li, J.-F.; Jiang, Y.; Hou, Y.-S.; Chen, C.; Peng, X.; Wang, Z. Chem. Asian J. 2017, 12, 2675. doi: 10.1002/asia.201701163
Xiao, F.-H.; Tian, J.-X.; Xing, Q.-Y.; Huang, H.-W.; Deng, G.-J.; Liu, Y.-J. ChemistrySelect 2017, 2, 428. doi: 10.1002/slct.201601549
(a) Shanmugapriya, J.; Rajaguru, K.; Muthusubramanian, S.; Bhuvanesh, N. Eur. J. Org. Chem. 2016, 2016, 1963.
(b) Huang, W.; Yang, G.-F. Bioorg. Med. Chem. 2006, 14, 8280.
Kong, D.-L.; Huang, T.; Liang, M.; Wu, M.-S.; Lin, Q. Org. Biomol. Chem. 2019, 17, 830. doi: 10.1039/C8OB02800A
Fan, W.; Chen, K.-Y.; Chen, Q.-P.; Li, G.-G.; Jiang, B. Org. Biomol. Chem. 2017, 15, 6493. doi: 10.1039/C7OB01515A
(a) Liu, Y.; Badsara, S. S.; Liu, Y.; Lee, C. RSC Adv. 2015, 5, 44299.
(b) Devi, N.; Rahaman, R.; Sarma, K.; Khan, T.; Barman, P. Eur. J. Org. Chem. 2017, 2017, 1520.
(c) Rafique, J.; Saba, S.; Rosrio, A. R.; Braga, A. L. Chem. Eur. J. 2016, 22, 79.
(d) Ji, X.-M.; Zhou, S.-J.; Chen, F.; Zhang, X.-G.; Tang, R.-Y. Synthesis 2015, 47, 659.
Rodrigues, J.; Saba, S.; Joussef, A. C.; Rafique, J.; Braga, A. L. Asian J. Org. Chem. 2018, 5, 1819.
Hazarika, S.; Gogoi, P.; Barman, P. RSC Adv. 2015, 5, 25765. doi: 10.1039/C5RA00677E
Kawashima, H.; Yanagi, T.; Wu, C.-C.; Nogi, K.; Yorimitsu, H. Org. Lett. 2017, 19, 4552. doi: 10.1021/acs.orglett.7b02147
Hostier, T.; Ferey, V.; Ricci, G.; Pardo, D. G.; Cossy, J. Org. Lett. 2015, 17, 3898. doi: 10.1021/acs.orglett.5b01889
Nalbandian, C. J.; Brown, Z. E.; Alvarez, E.; Gustafson, J. L. Org. Lett. 2018, 20, 3211. doi: 10.1021/acs.orglett.8b01066
Böhm, M. J.; Golz, C.; Rüter, I.; Alcarazo, M. Chem.-Eur. J. 2018, 24, 15026. doi: 10.1002/chem.201802806
(a) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359.
(b) Meanwell, N. A. J. Med. Chem. 2011, 54, 2529.
(c) Wang, J.; Sánchez-Roselló, M.; Aceña, J.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432.
(d) Landelle, G.; Panossian, A.; Leroux, F. R. Curr. Top. Med. Chem. 2014, 14, 941.
(a) Toulgoat, F.; Alazet, S.; Billard, T. Eur. J. Org. Chem. 2014, 2014, 2415.
(b) Shao, X.-X.; Xu, C.-F.; Lu, L.; Shen, Q. Acc. Chem. Res. 2015, 48, 1227.
(c) Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731.
(d) Chachignon, H.; Cahard, D. Chin. J. Chem. 2016, 34, 445.
Jereb, M.; Gosak, K. Org. Biomol. Chem. 2015, 13, 3103. doi: 10.1039/C4OB02633K
Horvat, M.; Jereb, M.; Iskra, J. Eur. J. Org. Chem. 2018, 2018, 3837. doi: 10.1002/ejoc.201800551
Bonazaba Milandou, L. J. C.; Carreyre, H.; Alazet, S.; Greco, G.; Martin-Mingot, A.; Ouamba, J.-M.; Bouazza, F.; Billard, T.; Thibaudeau, S. Angew. Chem., Int. Ed. 2017, 56, 169. doi: 10.1002/anie.201609574
Liu, S.; Zeng, X.; Xu, B. Asian J. Org. Chem. 2019, 8, 1372. doi: 10.1002/ajoc.201900358
Lu, S.; Chen, W.; Shen, Q. Chin. Chem. Lett. 2019, 30, 2279.
Zhang, P.; Li, M.; Xue, X.-S.; Xu, C.; Zhao, Q.; Liu, Y.; Wang, H.; Guo, Y.; Lu, L.; Shen, Q. J. Org. Chem. 2016, 81, 7486. doi: 10.1021/acs.joc.6b01178
Wang, D.; Zhang, R.; Lin, S.; Yan, Z.; Guo, S. M. RSC Adv. 2015, 5, 108030. doi: 10.1039/C5RA24351C
Xiao, F.; Chen, S.; Tian, J.; Huang, H.; Liu, Y.; Deng, G. Green Chem. 2016, 18, 1538. doi: 10.1039/C5GC02292D
Xu, Z.; Lu, G.; Cai, C. Org. Biomol. Chem. 2017, 15, 2804. doi: 10.1039/C6OB02823C
Lin, Y.-M.; Lu, G.-P.; Wang, G.-X.; Yi, W.-B. Adv. Synth. Catal. 2016, 358, 4100. doi: 10.1002/adsc.201600846
Yan, Q.; Jiang, L.; Yi, W.-B.; Liu, Q.; Zhang, W. Adv. Synth. Catal. 2017, 359, 2471. doi: 10.1002/adsc.201700270
Huang, Z.; Matsubara, O.; Jia, S.; Tokunaga, E.; Shibata, N. Org. Lett. 2017, 19, 934.
(a) Zhao, X.; Wei, A.; Yang, B.; Li, T.; Li, Q.; Qiu, D.; Lu, K. J. Org. Chem. 2017, 82, 9175.
(b) Zhao, X.; Zheng, X.; Tian, M.; Sheng, J.; Tong, Y.; Lu, K. Tetrahedron 2017, 73, 7233.
Chachignon, H.; Maeno, M.; Kondo, H.; Shibata, N.; Cahard, D. Org. Lett. 2016, 18, 2467. doi: 10.1021/acs.orglett.6b01026
Liu, J.; Zhao, X.; Jiang, L.; Yi, W.-B. Adv. Synth. Catal. 2018, 360, 4012. doi: 10.1002/adsc.201800702
Fernandez-Salas, J.; Pulis, A.; Procter, D. J. Chem. Commun. 2016, 52, 12364. doi: 10.1039/C6CC07627K
Chen, D.; Feng, Q.; Yang, Y.; Cai, X.; Wang, F.; Huang, S. Chem. Sci. 2017, 8, 1601. doi: 10.1039/C6SC04504A
(a) Yang, X.; Yan, R. Org. Biomol. Chem. 2017, 15, 3571.
(b) Wang, T.; Yang, F.; Tian, S. Adv. Synth. Catal. 2015, 357, 928.
(c) Rahaman, R.; Devi, N.; Sarmaa, K.; Barman, P. RSC Adv. 2016, 6, 10873.
(d) Bagdi, A. K.; Mitra, S.; Ghosh, M.; Hajra, A. Org. Biomol. Chem. 2015, 13, 3314.
(e) Rong, G.; Mao, J.; Yan, H.; Zheng, Y.; Zhang, G. J. Org. Chem. 2015, 80, 4697.
Pang, X.; Xiang, L. K.; Yang, X. D.; Yan, R. L. Adv. Synth. Catal. 2016, 358, 321. doi: 10.1002/adsc.201500943
Zhao, X.; Li, T. J.; Zhang, L. P.; Lu, K. Org. Biomol. Chem. 2016, 14, 1131. doi: 10.1039/C5OB02193F
Zhao, X.; Deng, Z. J.; Wei, A. Q.; Li, B. Y.; Lu, K. Org. Biomol. Chem. 2016, 14, 7304. doi: 10.1039/C6OB01006G
(a) Wang, D. Y.; Guo, S. M.; Zhang, R. X.; Lin, S.; Yan, Z. H. RSC Adv. 2016, 6, 54377.
(b) Wang, D. Y.; Zhang, R. X.; Lin, S.; Deng, R. H.; Yan, Z. H. Chin. J. Org. Chem. 2016, 36, 2757(in Chinese).
(王丁意, 张荣兴, 林森, 邓瑞红, 严兆华, 有机化学, 2016, 36, 2757.)
Li, J.; Zhu, D.; Lv, L.; Li, C.-J. Chem. Sci. 2018, 9, 5781. doi: 10.1039/C8SC01669K
Liu, P.; Liu, W.; Li, C.-J. J. Am. Chem. Soc. 2017, 139, 14315. doi: 10.1021/jacs.7b08685
(a) Wadman, M. Nature 2006, 440, 277
(b) Williams, R. B.; Norris, A.; Slebodnick, C.; Merola, J.; Miller, J. S.; Andriantsiferana, R.; Rasamison, V. E.; Kingston, D. G. J. Nat. Prod. 2005, 68, 1371.
(c) Waser, J.; Gaspar, B.; Nambu, H.; Carreira, E. M. J. Am. Chem. Soc. 2006, 128, 11693.
(d) Lin, Y.-M.; Lu, G.-P.; Wang, R.-K.; Yi, W.-B. Org. Lett. 2016, 18, 6424.
Zhao, W.; Xie, P.; Bian, Z.; Zhou, A.; Ge, H.; Niu, B.; Ding, Y. RSC Adv. 2015, 5, 59861. doi: 10.1039/C5RA10763F
Ding, Y.; Zhao, W.; Li, Y.; Xie, P.; Wu, W.; Zhou, A.; Huang, Y.; Liu, Y. Org. Biomol. Chem. 2016, 14, 1428. doi: 10.1039/C5OB02073E
Guo, T. Synth. Commun. 2017, 47, 2053. doi: 10.1080/00397911.2017.1364766
Zhao, W.; Xie, P.; Bian, Z.; Zhou, A.-H.; Ge, H.-B.; Zhang, M.; Ding, Y.; Zheng, L. J. Org. Chem. 2015, 80, 9167. doi: 10.1021/acs.joc.5b01602
Liu, W.-J.; Wang, S.-H.; Cai, Z.-H.; Li, Z.-Y.; Liu, J.-W.; Wang, A.-D. Synlett 2018, 29, 116. doi: 10.1055/s-0036-1588549
Wan, J.-P.; Zhong, S.; Xie, L.; Cao, X.; Liu, Y.; Wei, L. Org. Lett. 2016, 18, 584.
Bai, F.-C.; Zhang, S.; Wei, L.; Liu, Y.-Y. Asian J. Org. Chem. 2018, 7, 371. doi: 10.1002/ajoc.201700677
Siddaraju, Y.; Prabhu, K. R. J. Org. Chem. 2017, 82, 3084. doi: 10.1021/acs.joc.7b00073
Xiao, F.-H.; Wang, D.; Yuan, S.; Huang, H.; Deng, G.-J. RSC Adv. 2018, 8, 23319.
Fu, H.; Zhao, B.-T.; Zhu, W.-M. Tetrahedron Letters 2019, 60, 124. doi: 10.1016/j.tetlet.2018.11.072
Yang, F.-L.; Gui, Y.; Yu, B.-K.; Jin, Y.-X.; Tian, S.-K. Adv. Synth. Catal. 2016, 358, 3368. doi: 10.1002/adsc.201600795
(a) Bao, Y.; Yang, X.-Q.; Zhou, Q.-F.; Yang, F. L.; Org. Lett. 2018, 20, 1966.
(b) Bao, Y.; Zhong, L.-Y.; Hou, Q.; Zhou, Q.-F.; Yang, F.-L. Chin. J. Chem. 2018, 36, 1063.
Deng, L.-L.; Liu, Y.-Y. ACS Omega 2018, 3, 11890. doi: 10.1021/acsomega.8b01946
Guo, T.; Wei, X.-N. Synlett 2017, 28, 2499. doi: 10.1055/s-0036-1589083
Yang, Z.; Yan, Y.; Li, A.; Liao, J.; Zhang, L.; Yang, T.; Zhou, C. New J. Chem. 2018, 42, 14738. doi: 10.1039/C8NJ03461C
Dong, Y.-T.; Jin, Q.; Zhou, L.; Chen, J. Org. Lett. 2016, 18, 5708. doi: 10.1021/acs.orglett.6b02939
Bu, M.; Lu, G.; Cai, C. Org. Chem. Front. 2017, 4, 266. doi: 10.1039/C6QO00622A
Li, G.; Zhang, G.; Deng, X.; Qu, K.; Wang, H.; Wei, W.; Yang, D. Org. Biomol. Chem. 2018, 16, 8015.

Scheme 12 C—H trifluoromethylthiolation with N-trifluoro- methylthio-dibenzenesulfonimide

Scheme 15 Phosphine reagent catalyzed C—H fluoromethylthiolation with sodium fluoroalkylsulfinate

Scheme 16 Tf2O promoted C—H trifluoromethylthiolation with sodium trifluoroalkylsulfinate

Scheme 20 Possible mechanism of iodine-catalysed sulfenylation with sulfonyl hydrazides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:



























