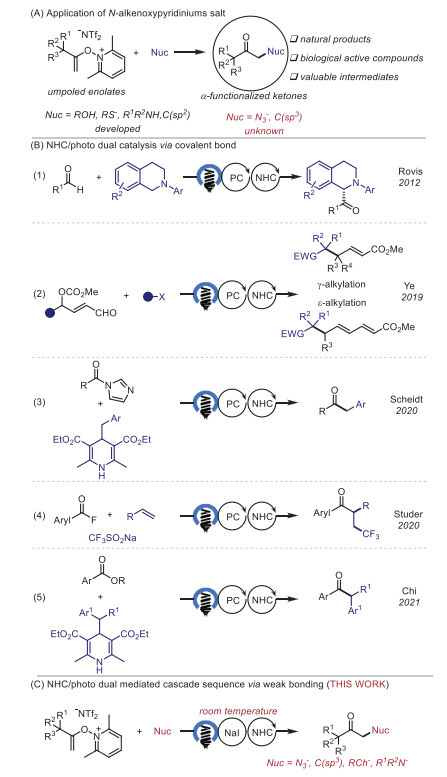
Scheme 1.
Motivation and synthetic strategy.
Visible-light-induced N-heterocyclic carbene mediated cascade transformation of N-alkenoxypyridinium salts
He Sheng , Qiang Liu , Fei Chen , Zhixiang Wang , Xiangyu Chen

α-Functionalized ketones are present in a wide range of bioactive compounds and have been proven to be versatile building blocks in organic synthesis (Scheme 1A) [1, 2]. Thus, tremendous efforts have been devoted to the development of novel methodologies for their synthesis. Umpolung is a fundamental concept in organic chemistry that provides an effective tool for synthetically important bond formations [3-6]. In recent years, the umpolung reactions of N-alkenoxypyridinium salts have emerged as a promising alternative strategy for the preparation of α-functionalized carbonyl compounds. Seminal studies by Gong [7], Hashimi [8], and Zhang [9] have established the synthetic utility of this approach for the synthesis of benzo-fused cyclic ketones and lactones via intramolecular reactions. In 2017, Xu and co-workers successfully employed N-alkenoxypyridinium salts in intermolecular reactions for the preparation of α-oxygenated ketones, α-thioketones, and α-thio thioketals [10]. Since then, this strategy has been extended to synthesize various α-functionalized ketones by using different nucleophiles [11-17]. Despite these successes, limited nucleophiles or high temperatures were generally required for these transformations, and the development of new activation modes allowing the wider scope of nucleophiles at room temperature is thus highly desirable. If realizable, such an approach is expected to increase the diversity of α-functionalized ketones and benefit the synthetic applications of N-alkenoxypyridinium salts.
In recent years, there has been a growing interest in using N-heterocyclic carbene (NHC)/photo dual catalysis to achieve new activation modes for the development of alternative methods for the synthesis of useful compounds (Scheme 1B) [18-29]. In 2012, Rovis and co-workers first reported the α-acylation of tertiary amines with aldehydes demonstrating the ability of NHC/photoredox dual catalysis [30]. In 2019, Ye, Gao and co-workers developed an elegant example of γ- and ε-alkylation of dienolates via merging of photoredox and NHC catalysis [31-34]. The NHC/photo dual catalyzed transformations of acyl azoliums were developed by research groups of Scheidt [35, 36] and Studer [37-39], respectively. The photocatalyst-free, direct photoexcitation of acyl azoliums was also realized by Chi and co-workers [40]. However, all of these transformations are based on the NHC-based intermediates via covalent bonds. In our previous study, we found that the electrostatic interaction of NHC and alkali metal was enabled to create a photoactive electron donor-acceptor (EDA) complex for the iodination of N-alkenoxypyridiniums [41]. As part of our ongoing research in weak interaction enabled photosynthesis [29, 41-44], we set out to explore the possibility of developing an NHC-mediated functionalization of N-alkenoxypyridinium salts with various nucleophiles, including tetramethylammonium azide, secondary amines, aryl and alkyl thiols, and even the challenging C(sp3)-nucleophiles (Scheme 1C).
To validate the feasibility of the proposed process, we first targeted the azidation of 1 with tetramethylammonium azide under NHC catalysis for the synthesis of α-azido ketones (Table 1), which are very versatile and valuable synthetic intermediates [45, 46]. We were pleased to find that the reaction went well in the presence of NHC A as the precatalyst and LiNTf2 as an ion exchange additive to increase the solubility of NHC salt, under irradiation of blue light, and gave the desired product 2 in 78% yield (Table 1, entry 1). The screening of the influence of solvent showed that MeCN was the best choice (entry 2). Other NHCs, such as B, C, D, E and F, were evaluated but gave inferior results (entry 3), and the reactivity was decreased without NHC, LiNTf2 or NaI (entries 4–6). Control experiments confirmed the necessity of visible light (entry 7).
With the optimized conditions in hand, we subsequently examined the generality of the method (Scheme 2). We successfully converted a range of unfunctional alkenoxypyridiniums into the corresponding α-azido ketones with acyclic and cyclic alkyl groups (2–7). This was also true for the substrates bearing aryl groups, providing the products 8–11 without an apparent change in the yields. The substrates with various functional groups, such as dimethyl malonate (12), esters (13–16), amide (17), ethers (18–20) also proceeded well and afforded the desired products in 41%−90% yield. In addition, a substrate containing a complex natural product skeleton (estrone) also gave good yield (54%) of azide product 21.
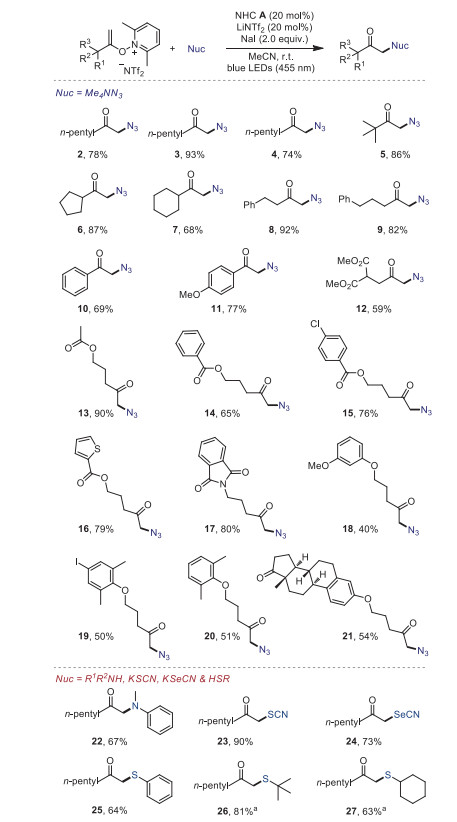
Encouraged by these results, we then turned our attention to other α-functionalized ketones. α-Amino ketone, α-carbonyl selenocyanates, thiocyanates and α-thioketones are common structural motifs found in many biologically active compounds. We were pleased to find that all of these types of compounds (22−27) can be got in good to high yields by using our developed protocol at room temperature. Notably, it is unable to get the α-alkyl thioketones by using the traditional method under heat conditions [16].
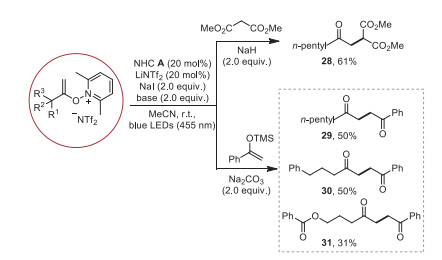
The construction of the C(sp3)-C(sp3) bond by using N-alkenoxypyridinium salts is a continued challenge and cannot be achieved under traditional heat conditions. Gratifyingly, the reaction of N-alkenoxypyridinium salt and dimethyl malonate could react smoothly and successfully constructed C(sp3)-C(sp3) bond (Scheme 3, 28). Moreover, the 1, 4-dicarbonyl compounds, which are frequently found in many natural products and bioactive compounds [47-49], can be easily obtained from the reaction of trimethyl((1-phenylvinyl)oxy)silane and N-alkenoxypyridinium salts (Scheme 3, 29–31).
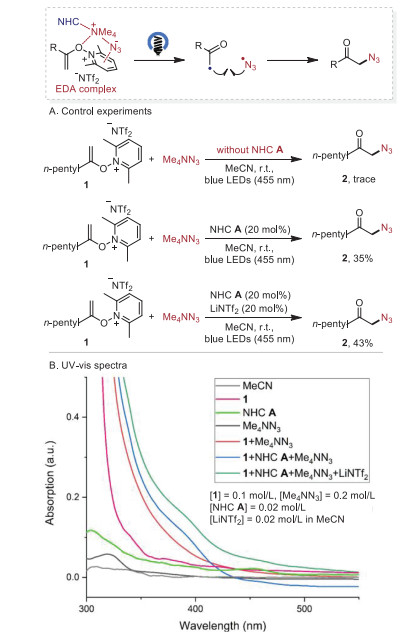
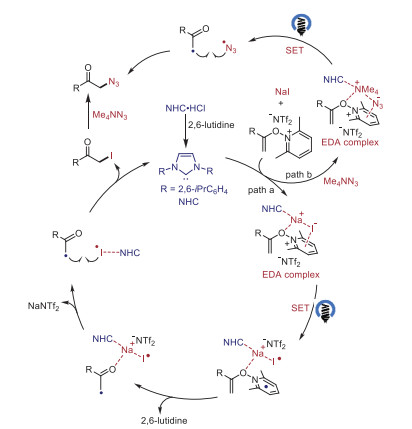
A series of experiments were carried out to clarify the possible reaction pathways (Scheme 4). Pathway A, involving the formation of α-iodo ketone followed by nucleophilic substitution of azide anion was possible by the control experiments in which the reaction of N-alkenoxypyridinium salt 1 and NaI under the reaction conditions gave the α-iodo ketone in 92% yield and the α-iodo ketone reacted well with tetramethylammonium azide to afford the desired product 2 in 95% yield (Scheme 4A). Furthermore, we studied the UV–vis absorption spectra of the reaction. While neither N-alkenoxypyridinium salt 1 nor NaI showed significant UV–vis absorption in the visible region of the spectra, an obvious red shift of absorption was observed in the spectrum of the reaction mixture 1 and NaI, supporting the formation of an EDA complex between them. The addition of NHC A further enhanced the red shift of absorption, thus facilitating the EDA complex formation (Scheme 4B). In addition, the Benesi−Hildebrand analysis of 1 and NaI complex provided an association constant (KEDA) of 8.1 L/mol in MeCN and the addition of NHC A improved the KEDA to 16.1 L/mol (Scheme 4C).

Pathway B involves the formation of an EDA complex with N-alkenoxypyridinium salt 1, tetramethylammonium azide and NHC A followed by cross-coupling of the resultant alkyl radical and azide radical (Scheme 5). While only trace amounts of the desired product 2 were observed for the reaction of N-alkenoxypyridinium salt 1 and tetramethylammonium azide. The addition of NHC A, and NHC A/LiNTf2 led to 35% and 43% yield, respectively (Scheme 5A). The UV–vis absorption spectra studies revealed the formation of EDA complex among the N-alkenoxypyridinium salt 1, tetramethylammonium azide, LiNTf2 and NHC A, where the addition of NHC A or NHC A/LiNTf2 caused an obvious red shift of absorption (Scheme 5B).
On the basis of our previous studies [29, 41] and above studies, a possible mechanism for the α-functionalized ketones formation process is outlined in Scheme 6. The EDA complex between the N-alkenoxypyridinium salt and NaI is further activated by complexation with an NHC. Irradiation of this proposed ternary complex generates the alkyl radical, iodine radical, and 2, 6-lutidine. The following radical−radical coupling gives the iodination product which could be attacked by the nucleophiles to afford the final α-functionalized ketones. Concomitantly, the NHC is formed via deprotonation of the corresponding azolium salt by the in situ generated 2, 6-lutidine. In this process, the 2c−3e interaction of NHC could stabilize the generated iodine radical, thus improving the efficiency of the iodinations. Notably, the reaction may also involve the less effective cross-coupling of alkyl radical and azide radical (path B).
In summary, an NHC-mediated cascade radical-radical coupling/nucleophilic substitution sequence of N-alkenoxypyridinium salts with various nucleophiles has been successfully developed. The corresponding α-functionalized ketones, including α-azido ketones, α-amino ketone, α-carbonyl selenocyanates, thiocyanates and α-thioketones, were obtained in moderate to good yields. In addition, the challenging C(sp3)-C(sp3) bond formation reaction was also successful by using this methodology. We anticipate that the presented synthetic method could be applied in the pharmaceutical and organic chemistry arenas.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
We acknowledge financial support from the National Natural Science Foundation of China (No. 22001248) and the Fundamental Research Funds for the Central Universities and the University of the Chinese Academy of Sciences.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.01.028.
W. Notz, F. Tanaka, C.F. Barbas, Acc. Chem. Res. 37 (2004) 580–591. doi: 10.1021/ar0300468
S. Mukherjee, J.W. Yang, S. Hoffmann, B. List, Chem. Rev. 107 (2007) 5471–5569. doi: 10.1021/cr0684016
D. Seebach, Angew. Chem. Int. Ed. 18 (1979) 239–258. doi: 10.1002/anie.197902393
X. Bugaut, F. Glorius, Chem. Soc. Rev. 41 (2012) 3511–3522. doi: 10.1039/c2cs15333e
A. Grossmann, D. Enders, Angew. Chem. Int. Ed. 51 (2012) 314–325. doi: 10.1002/anie.201105415
M.H. Wang, K.A. Scheidt, Angew. Chem. Int. Ed. 55 (2016) 14912–14922. doi: 10.1002/anie.201605319
D.F. Chen, Z.Y. Han, Y.P. He, J. Yu, L.Z. Gong, Angew. Chem. Int. Ed. 51 (2012) 12307–12310. doi: 10.1002/anie.201205062
K. Graf, C.L. Rühl, M. Rudolph, F. Rominger, A.S.K. Hashmi, Angew. Chem. Int. Ed. 52 (2013) 12727–12731. doi: 10.1002/anie.201304813
Z. Xu, H. Chen, Z. Wang, A. Ying, L. Zhang, J. Am. Chem. Soc. 138 (2016) 5515–5518. doi: 10.1021/jacs.6b02533
Z. Xu, R. Zhai, T. Liang, L. Zhang, Chem. Eur. J. 23 (2017) 14133–14137. doi: 10.1002/chem.201703087
Q. Liu, C.S. Zhang, H. Sheng, et al., Org. Lett. 22 (2020) 5617–5621. doi: 10.1021/acs.orglett.0c01984
G.R. Mathi, B. Kweon, Y. Moon, Y. Jeong, S. Hong, Angew. Chem. Int. Ed. 59 (2020) 22675–22683. doi: 10.1002/anie.202010597
N. Wu, C. Li, J. Mi, Y. Zheng, Z. Xu, Org. Lett. 22 (2020) 7118–7122. doi: 10.1021/acs.orglett.0c02457
X. Tang, N. Wu, R. Zhai, et al., Org. Biomol. Chem. 17 (2019) 966–972. doi: 10.1039/C8OB02907E
X. Xia, B. Chen, X. Zeng, B. Xu, Org. Biomol. Chem. 16 (2018) 6918–6922. doi: 10.1039/C8OB02004C
X. Xia, B. Chen, X. Zeng, B. Xu, Adv. Synth. Catal. 360 (2018) 4429–4434. doi: 10.1002/adsc.201800968
R.L. Zhai, Y.S. Xue, T. Liang, J.J. Mi, Z. Xu, J. Org. Chem. 83 (2018) 10051–10059. doi: 10.1021/acs.joc.8b01388
Y. Wen, H. Weimin, D. Xiuqin, L. Xin, S. Jianwei, Angew. Chem. Int. Ed. 55 (2016) 15783–15786. doi: 10.1002/anie.201608371
L. Su, H. Sun, J. Liu, C. Wang, Org. Lett. 23 (2021) 4662–4666. doi: 10.1021/acs.orglett.1c01400
H. Ohmiya, ACS Catal. 10 (2020) 6862–6869. doi: 10.1021/acscatal.0c01795
A. Mavroskoufis, M. Jakob, M.N. Hopkinson, ChemPhotoChem 4 (2020) 5147–5153. doi: 10.1002/cptc.202000120
Y. Sumida, H. Ohmiya, Chem. Soc. Rev. 50 (2021) 6320–6332. doi: 10.1039/D1CS00262G
L. Marzo, Eur. J. Org. Chem. 2021 (2021) 4603–4610. doi: 10.1002/ejoc.202100261
J. Liu, X.N. Xing, J.H. Huang, L.Q. Lu, W.J. Xiao, Chem. Sci. 11 (2020) 10605–10613. doi: 10.1039/D0SC03595E
K.Q. Chen, H. Sheng, Q. Liu, P.L. Shao, X.Y. Chen, Sci. China Chem. 64 (2021) 7–16. doi: 10.1007/s11426-020-9851-8
Y.Y. Liu, J. Liu, L.Q. Lu, W.J. Xiao, Top. Curr. Chem. 377 (2019) 37. doi: 10.1007/s41061-019-0265-0
T. Ishii, K. Nagao, H. Ohmiya, Chem. Sci. 11 (2020) 5630–5636. doi: 10.1039/D0SC01538E
L. Dai, S. Ye, Chin. Chem. Lett. 32 (2021) 660–667. doi: 10.1016/j.cclet.2020.08.027
K.Q. Chen, Z.X. Wang, X.Y. Chen, Org. Lett. 22 (2020) 8059–8064. doi: 10.1021/acs.orglett.0c03006
D.A. DiRocco, T. Rovis, J. Am. Chem. Soc. 134 (2012) 8094–8097. doi: 10.1021/ja3030164
Z.H. Gao, Z.H. Xia, L. Dai, S. Ye, Adv. Synth. Catal. 362 (2020) 1819–1824. doi: 10.1002/adsc.202000164
Z.H. Xia, L. Dai, Z.H. Gao, S. Ye, Chem. Commun. 56 (2020) 1525–1528. doi: 10.1039/C9CC09272B
L. Dai, Z.H. Xia, Y.Y. Gao, Z.H. Gao, S. Ye, Angew. Chem. Int. Ed. 58 (2019) 18124–18130. doi: 10.1002/anie.201909017
L. Dai, S. Ye, Org. Lett. 22 (2020) 986–990. doi: 10.1021/acs.orglett.9b04533
A.V. Bay, K.P. Fitzpatrick, R.C. Betori, K.A. Scheidt, Angew. Chem. Int. Ed. 59 (2020) 9143–9148. doi: 10.1002/anie.202001824
A.V. Bay, K.P. Fitzpatrick, G.A. González-Montiel, et al., Angew. Chem. Int. Ed. 60 (2021) 17925–17931. doi: 10.1002/anie.202105354
Q.Y. Meng, N. Döben, A. Studer, Angew. Chem. Int. Ed. 59 (2020) 19956–19960. doi: 10.1002/anie.202008040
Q.Y. Meng, L. Lezius, A. Studer, Nat. Commun. 12 (2021) 2068. doi: 10.1038/s41467-021-22292-z
K. Liu, A. Studer, J. Am. Chem. Soc. 143 (2021) 4903–4909. doi: 10.1021/jacs.1c01022
S.C. Ren, W.X. Lv, X. Yang, et al., ACS Catal. 11 (2021) 2925–2934. doi: 10.1021/acscatal.1c00165
H. Sheng, Q. Liu, X.D. Su, et al., Org. Lett. 22 (2020) 7187–7192. doi: 10.1021/acs.orglett.0c02523
K.Q. Chen, J. Shen, Z.X. Wang, X.Y. Chen, Chem. Sci. 12 (2021) 6684–6690. doi: 10.1039/D1SC01024G
Q. Liu, Y. Lu, H. Sheng, et al., Angew. Chem. Int. Ed. 60 (2021) 25477–25484. doi: 10.1002/anie.202111006
Q. Liu, X.Y. Chen, Org. Chem. Front. 7 (2020) 2082–2087. doi: 10.1039/D0QO00494D
T. Patonay, K. Kónya, É. Juhász-Tóth, Chem. Soc. Rev. 40 (2011) 2797–2847. doi: 10.1039/c0cs00101e
S. Faiz, A.F. Zahoor, N. Rasool, et al., Molecules 20 (2015) 14699–14745. doi: 10.3390/molecules200814699
M.P. DeMartino, K. Chen, P.S. Baran, J. Am. Chem. Soc. 130 (2008) 11546–11560. doi: 10.1021/ja804159y
T. Fujisawa, K. Igeta, S. Odake, et al., Bioorg. Med. Chem. 10 (2002) 2569–2581. doi: 10.1016/S0968-0896(02)00109-8
M. Whittaker, C.D. Floyd, P. Brown, A.J.H. Gearing, Chem. Rev. 99 (1999) 2735–2776. doi: 10.1021/cr9804543

Scheme 2 Reaction scope. Yields of isolated products are given. a 2.0 equiv. of K2CO3 was added.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:



