Citation:
Xue Xin, Qiming Qu, Islam E. Khalil, Yuting Huang, Mo Wei, Jie Chen, Weina Zhang, Fengwei Huo, Wenjing Liu. Hetero-phase zirconia encapsulated with Au nanoparticles for boosting electrocatalytic nitrogen reduction[J]. Chinese Chemical Letters,
2024, 35(5): 108654.
doi:
10.1016/j.cclet.2023.108654
Hetero-phase zirconia encapsulated with Au nanoparticles for boosting electrocatalytic nitrogen reduction
English
Hetero-phase zirconia encapsulated with Au nanoparticles for boosting electrocatalytic nitrogen reduction
Key Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Nanjing Tech University, Nanjing 211816, China
* Corresponding author. E-mail address: iamwjliu@njtech.edu.cn (W. Liu). 1 These authors contributed equally to this work.
Received Date:
03 May 2023 Accepted Date:
05 June 2023 Revised Date:
24 May 2023 Available Online:
15 May 2024
Abstract:
Electrocatalytic nitrogen reduction reaction (NRR) is considered as an attractive approach for ammonia synthesis under mild conditions. A bottleneck of NRR is the exploration of efficient catalysts for accelerating reaction kinetics, among which heterogeneous structures possessing distinct atomic arrangement could modify electronic structure, and therefore altering their NRR activity. Here, we report a facile strategy for fabricating hetero-phase metal oxides derived from metal organic framework that are further integrated with Au nanoparticles as NRR catalysts. The phase composition of zirconia can be easily adjusted by simply changing the reaction temperature, where the monoclinic and tetragonal phases with the roughly close proportions have a distinct interface, leading to a strong interaction between Au and ZrO2. The enhanced interaction renders Au to be more electropositive and facilitates stronger binding to N2. As a result, a remarkable ammonia yield of 22.32 µg h−1 mgcat.−1 and a Faradaic efficiency of 31.92% can be achieved at low overpotential. This work is expected to pave the way for the design of heterogeneous structures and the exploration of hetero-phase nanostructures in boosting the electrocatalytic NRR.
The production of ammonia (NH3) through the reduction of nitrogen gas (N2) at ambient conditions is a topic of significant interest. The traditional Haber-Bosch process, which has been developed over a century, demands for high pressure (> 10 MPa) and temperature (> 300 ℃) to produce NH3. This process emits a large amount of carbon footprints and consumes intensive energy [1–3]. Therefore, there has been a pursuit to find an alternative and greener way to produce NH3. One promising approach is the electrocatalytic reduction reaction (NRR), which replaces the steam reforming unit, and therefore lowers the energy consumption and the gas emission. This method brings about the following benefits: (1) converting alternative energy in rural parts to portable chemicals; (2) eliminating CO2 emissions; (3) providing a simpler system for localized and on-site/demand ammonia production; (4) presenting a modular technology that is advantageous to the imitation of electrochemical technique [4–7].
Thus far, the design of highly efficient electrocatalysts to overcome the indolence and stability of N2 molecule [8–10] and further enhance NRR efficiency has been a specific focus of the electrochemical reduction method [11–15]. Among them, hetero-phase structures possess a heterogeneous region between which could produce integrated characteristics caused by interaction coupling, which are over the prediction by the rule-of-mixtures. Hetero-phase structures applied to NRR include the following advantages: (1) Hetero-phase materials have a high proportion of atoms at their phase interfaces, resulting in a very high number of unsaturated bonds, suspended bonds, dislocations and defects, which are very conducive to creating catalytic active sites to accelerate the catalytic reaction [16]. (2) The heterogeneous zone could promote the contact between N2 and the catalyst, causing the enrichment of N2 concentration and the evacuation of proton concentration, thereby inhibiting the occurrence of hydrogen evolution reaction, which is a competition side reaction of NRR [17]. Moreover, such hetero-phase structures have been rarely reported for NRR yet.
Herein, we provide a facile strategy for synthesizing hetero-phase materials. By pyrolysis of metal−organic frameworks (MOFs) integrated with Au as the precursor, and controlling the pyrolysis temperature, Au/ZrO2 with heterogeneous monoclinic and tetragonal phase structure could be easily synthesized. The design of the as-synthesized material is mainly based on the following advantages: (1) MOFs are porous materials consisting of metal nodes linked by organic ligands, have been applied in electrocatalysis owing to their tailorable structure, high porosity, well-defined pore size distribution, high surface area, and low density [18–20]. The use of MOFs as precursors for preparing MOF derivatives for NRR as they can provide plenty of exposed active sites for electron transfer, N≡N cleavage, and N2 adsorption. (2) Au is rich in valence electrons and unoccupied orbitals, and has been arguably as one of the most promising electrocatalysts for NRR [17,21,22]. To make more efficient use of the precious Au, it is often dispersed onto an inert substrate [23–26]. Metal particles dispersed on support usually have a low coordination number and can serve as metal active centers, in addition, the metal particles own a strong coupling with the carrier, which can enhance the catalytic activity of gold-based catalysts. (3) Zirconium dioxide have been shown to inhibit hydrogen precipitation for efficient nitrogen fixation [27]. Moreover, ZrO2 has a multi-phase structure, and the interfaces created by the phase boundaries will introduce defects and facilitate electronic transportion. Among the templates for synthesizing ZrO2, MOFs play a vital role in forming the combining of monoclinic and tetragonal phases. The catalyst with integrated phases exhibits superior performance to its counterparts. Specifically, Au/ZrO2–800 catalyst achieves an ammonia yield of 22.32 µg h−1 mgcat.−1 and the corresponding FE of 16.93% at −0.1 V vs. reversible hydrogen electrode (RHE) in 0.1 mol/L KOH solution, with a highest Faradaic efficiency (FE) of 31.92% and the homologous ammonia yield of 13.63% at −0.04 V vs. RHE. This work has significant implications for exploring hetero-phase nanostructures in advancing the electrocatalytic NRR.
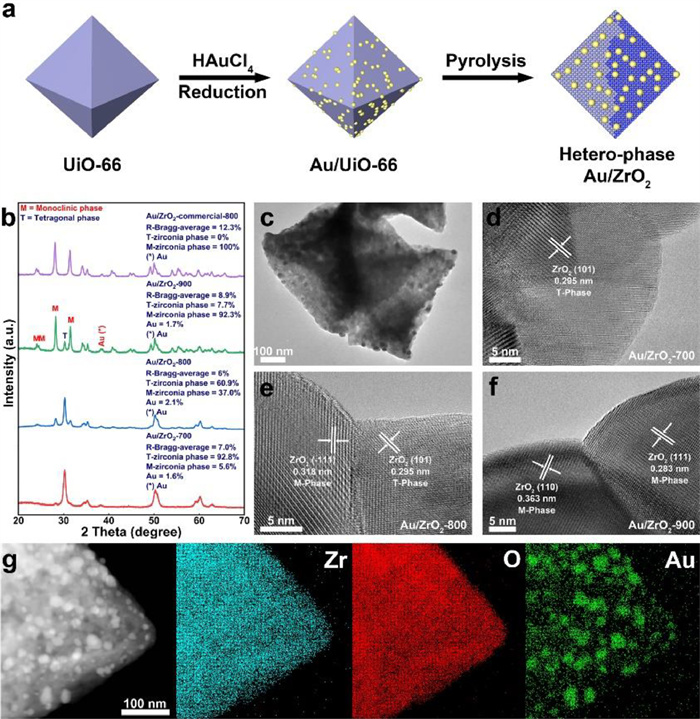
UiO-66 (Zr6O4(OH)4(BDC)6; BDC: 1, 4-benzenedicarboxylate), a kind of MOF, was synthesized first and then confined with Au ions by a hydrothermal method, followed by being annealed at 300 ℃ to form Au/UiO-66. Transmission electron microscopy (TEM) image shows that Au nanoparticles (NPs) with the size range of 4–7 nm are well distributed on the surface of UiO-66 (Fig. S1 in Supporting information). To verify the conversion process of UiO-66 to ZrO2, thermogravimetric analysis (TGA) measurement was adopted (Fig. S2 in Supporting information), which reveals two distinct mass drops. The first stage varying from 25 ℃ to 200 ℃ should be attributed to the evaporation of some small molecules such as water, while the second stage from 400 ℃ to 500 ℃ is caused by the degradation of the ligand and the demolition of the UiO-66 carbon framework. At around 530 ℃, the UiO-66 structure utterly collapses, and in fact, the practical calcination temperature was much higher than 530 ℃, which ensures the decomposition of UiO-66 and the formation of ZrO2. The weight loss calculated theoretically from UiO-66 to ZrO2 as the total molecular weight of UiO-66 (C48H24O30Zr6) is 1628.03 and after subtracting the molecular weight 600.72 of C and H atoms, it is found that the weight loss ratio 36.89% matches well with the experimental result of TGA curve. After tardily calcinating Au/UiO-66 at various temperature ranging from 700 ℃ to 900 ℃ under an Ar atmosphere for 3 h, Au/ZrO2-T with mixed-composition phases were obtained (Fig. 1a).
Figure 1
Figure 1.
(a) Scheme image of preparation of hetero-phase Au/ZrO2. (b) XRD patterns of hetero-phase Au/ZrO2. (c) TEM image of Au/ZrO2–800 composites. HRTEM images of (d) Au/ZrO2–700, (e) Au/ZrO2–800 and (f) Au/ZrO2–900. (g) STEM image and the corresponding EDX elemental mapping images of Au/ZrO2–800 composites.
As shown in Fig. 1b, X-ray diffraction (XRD) analysis indicates that the pyrolysis of Au/UiO-66 yield monoclinic/tetragonal hetero-phase Au/ZrO2. The XRD patterns of Au/ZrO2–700, Au/ZrO2–800, Au/ZrO2–900 derived from Au/UiO-66 at different temperatures and that of Au-ZrO2-commercial at 800 ℃ were compared and analyzed. The characteristic diffraction peaks of ZrO2 at 24.0°, 28.2°, 31.5° correspond respectively to the (110), (), and (111) lattice planes of monoclinic ZrO2 (PDF#86–1449), while those at 30.2° and 35.3°, are respectively attributed to the (101) and (200) lattice planes of tetragonal ZrO2 (PDF#88–1007). The ratio of phase transition is calculated based on the refinement of the XRD peaks by Rietveld [28]. The peak positions are adjusted by the successive refinement of systematic errors, taking into consideration zero-shifting and sample-shifting errors (Figs. S3-S6 in Supporting information). The tetragonal and monoclinic phase percentages for each catalyst are recapitulated in Table S1 (Supporting information). The successful synthesis of stable hetero-phase structure requires precise control of the carbonization temperature, as pyrolysis at 700 ℃, the main product obtained is tetragonal phase; while pyrolysis at high temperature of 900 ℃, the main stuff obtained is monoclinic phase; when carbonization is performed with the commerical ZrO2 loaded Au, a pure monoclinic phase of ZrO2 is obtained; only the temperature is controlled to 800 ℃, a considerable proportion of hetero-phase structure could be obtained.
Transmission electron microscopy (TEM) measurement was used to investigate the morphology (Fig. 1c). The phase locations in Au/ZrO2–700, 800, 900 catalysts were further analyzed by high-resolution TEM (HRTEM) in the fast Fourier transform (FFT) (Figs. 1d–f). The distinct lattice fringes of 0.283, 0.318, and 0.363 nm are accredited to (111), (), and (110) planes of monoclinic ZrO2, respectively, and 0.295 nm is identified to (101) plane of tetragonal ZrO2. The existence form of Au in Au/ZrO2–800 is also characterized, from HRTEM image in Fig. S7 (Supporting information), the distinct lattice fringe of 0.235 nm is accredited to (111) planes of Au NPs (PDF#99–0056). Scanning transmission electron microscopy energy dispersive X-ray spectroscopy (STEM-EDX) was employed for mapping analysis of Zr, O and Au elements distribution in Au/ZrO2–800 (Fig. 1g). It reveals that the Zr and O elements are homogeneously distributed over the selected area, while Au is mainly concentrated in the bright region, thus further confirming the existence of Au NPs in ZrO2.
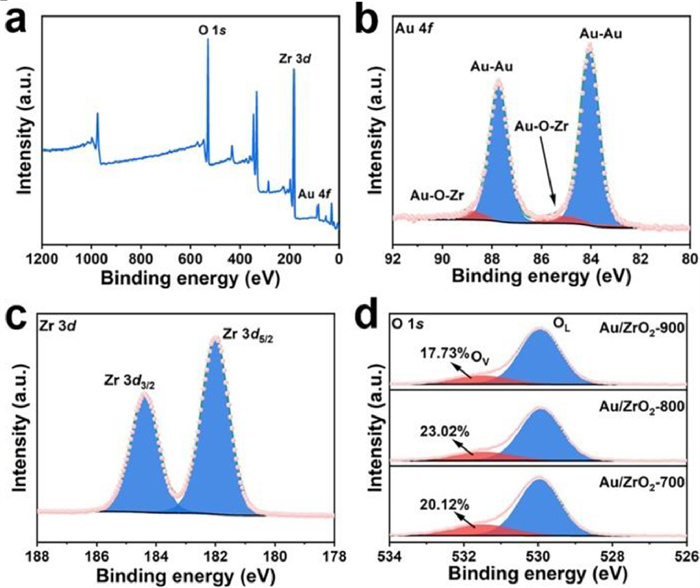
Further analysis of Au/ZrO2–800 by X-ray photoelectron spectroscopy (XPS) probes the chemical valence of the surface elements and the interaction between metal and hetero-phase carrier. XPS survey spectrum reveals that Au/ZrO2–800 is mainly composed of O, Zr and Au elements (Fig. 2a). In the fine spectrum of Au element, two pairs of spin-orbit doublets indicate the existence of Au0 (Au-Au bonding energy peaks at 84.0 and 87.7 eV) and Auδ+ (Au-O bonding energy peaks at 85.9 and 88.8 eV) (Fig. 2b), which is consistent with the previously published results [29,30]. Furthermore, in the fine spectrum of Zr (Fig. 2c), the binding energies at 182.0 eV and 184.4 eV can be ascribed to the characteristic peaks of Zr 3d5/2 and Zr 3d3/2, which is lower than the values of ZrO2 (182.8 and 185.3 eV) reported in the literature [31]. The presence of Auδ+ and the negative displacement of Zr4+ in this sample indicate the electron transfer from the metal particles to the oxide carrier, suggesting that the Au particles may form Au-O-Zr bonding with the ZrO2 carrier. In addition to strong-metal support interation, small sized metal particles own high surface energy, a strong chemical bonding between metal and the coordinated lattice oxygen of oxides is always formed in order to keep the metal stability [32–34]. The O 1s spectra of Au/ZrO2 annealed at different temperatures can be fitted to two peaks, i.e., lattice oxygen (OL) and oxygen vacancy (OV), and all peaks of OL and OV are located at 529.9 and 531.5 eV, respectively (Fig. 2d). OV is believed to promote N2 adsorption and activation in the NRR process, and therefore affect the NRR performance [35–39]. The OV contents of Au/ZrO2–700, 800, 900 can be estimated from the area ratio of the XPS fit as 20.12%, 23.02% and 17.73%, respectively, which indicates that when Au/ZrO2 is annealed, the interface forming at the phase boundary introduces defects, and Au/ZrO2 with roughly close proportions of mixed-composition phases can produce more OV.
Figure 2
Figure 2.
(a) XPS spectra of Au/ZrO2–800. The corresponding fine spectra of (b) Au 4f (c) Zr 3d and (d) O 1s.
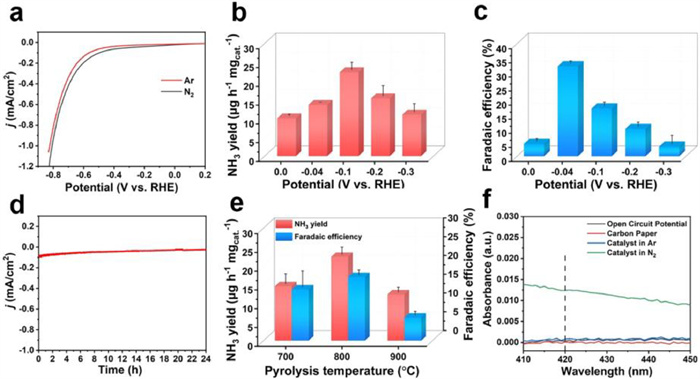
The electrocatalytic NRR performance was measured in H-type cell with N2-saturated 0.1 mol/L KOH as the electrolyte under ambient conditions, and all potentials were converted to the RHE scale (Fig. S8 in Supporting information). Electrolytes were collected at the end of the test, ammonia concentrations were measured plying Nessler's reagent spectrophotometry, and the concentration of the possible by-product hydrazine was determined utilizing the Watt and Chrisp method [40]. Calibration curves for the determination of ammonia and hydrazine were plotted respectively (Figs. S9 and S10 in Supporting information). The as-synthesized Au/ZrO2-T was then used as catalyst to study its NRR catalytic performance. To determine the potential of the electrochemically active region of NRR, linear-sweep voltammetric (LSV) curves were measured in both N2- and Ar-saturated electrolytes (Fig. 3a), which exhibit similar shapes. However, there is an increase in current density below 0.0 V in N2-saturated electrolyte, indicating the formation of NH3 from N2 at the reduction potential.
Figure 3
Figure 3.
Electrocatalytic NRR on Au/ZrO2–800 catalyst in 0.1 mol/L KOH electrolyte. (a) LSV curves in N2- and Ar-saturated electrolytes. (b) NH3 yield at different applied potentials. (c) FEs at diverse applied potentials. (d) Chronoamperometry test at –0.1 V for 24 h. (e) Comparison of electrocatalytic NRR on Au/ZrO2–700, Au/ZrO2–800 and Au/ZrO2–900 catalysts in 0.1 mol/L KOH electrolyte. (f) UV–vis absorption spectra under different conditions.
Furthermore, electrocatalytic tests were carried out on Au/ZrO2–800 catalyst at an given potential from 0.0 V to −0.3 V to study its catalytic activity and the optimal working potential for NRR. The current density increases markedly as the applied potential moves from 0.0 V to −0.3 V (Fig. S11 in Supporting information). After two hours of the electrolysis process, the electrolyte is collected and then quantified by Nessler's reagent to determine the NH3 yield and the corresponding Faradaic efficiency by formulae (see Supporting information). The highest NH3 yield with 22.32 µg h−1 mgcat.−1 is obtained at −0.1 V with a FE of 16.93%. The highest FE of 31.92% is obtained at −0.04 V with a NH3 yield of 13.63%, both high yield and high efficiency are achieved at very low voltages (Figs. 3b and c). The corresponding ultraviolet-visible (UV–vis) absorption curves of the electrolytes are presented in Fig. S12 (Supporting information). Simultaneously, a 24 h long-term electrocatalytic test was performed at −0.1 V. After electrolysis for 24 h, chronoamperometry curve of Au/ZrO2–800 shows negligible decrease in current density (Fig. 3d), reflecting the excellent stability of Au/ZrO2–800. In addition, the possible by-product hydrazine is not detected at each potential, indicating the excellent selectivity of Au/ZrO2–800 (Fig. S13 in Supporting information).
To investigate the importance of the hetero-phase structures for boosting NH3 performance, Au/ZrO2 with different ratios of monoclinic and tetragonal phases were prepared by controlling the pyrolysis temperature. In Table S1, Au/ZrO2–800 shows monoclinic and tetragonal phase ratios of 37% and 60.9%, respectively, which are relatively closer compared with other samples. Notably, MOFs play a vital role in forming the hetero-phase structures compared to other templates for synthesizing Au/ZrO2. The pure tetragonal phase of Au/ZrO2-commercial were achieved when we utilized the commercially purchased ZrO2 directly instead of MOF to load Au. The electrocatalytic NRR performances of samples Au/ZrO2-T were also tested at −0.1 V (Fig. 3e and Fig. S14 in Supporting information). Compared with Au/ZrO2–700 (12.50 µg h−1 mgcat.−1), Au/ZrO2–900 (12.32 µg h−1 mgcat.−1) and Au/ZrO2-commercial (0.95 µg h−1 mgcat.−1), Au/ZrO2–800 achieves the highest NH3 yield rate of 22.32 µg h−1 mgcat.−1 and the highest FE of 31.92%, indicating the closer ratios of the monoclinic phase and tetragonal phase, the higher performance the catalyst will achieve. Furthermore, in order to clarify whether the origin of the catalytic activity is due to the interaction between Au and the hetero-phase carrier, ZrO2-800 without introduction of metal was also designed. The NH3 yield of ZrO2–800 was tested at −0.1 V, which was only a quarter of that of Au/ZrO2–800 (Fig. S14).
To gain insights into the reasons behind the NRR performance differences, the electrochemically active surface area (ESCA) was further studied. The cyclic voltammetry (CV) curves of Au/ZrO2–700, 800, 900 in N2-saturated 0.1 mol/L KOH electrolyte were tested. The double-layer capacitance (Cdl) of the catalytic surface was measured by CV in a Faradaic silence potential range of 0.02 ∼ −0.08 V vs. Ag/AgCl using different scan rates, which is generally used to represent the corresponding ESCA. As shown in Fig. S15 (Supporting information), Au/ZrO2–800 exhibits a higher Cdl value of 0.0648 mF/cm2 than Au/ZrO2–700, 900 of 0.0585 mF/cm2 and 0.0495 mF/cm2, respectively, indicating that the integration of the monoclinic and tetragonal phases in Au/ZrO2–800 can enhance the ESCA effectively.
To investigate if the detected NH3 is a result of the electrocatalytic NRR process, control experiments were performed (Fig. 3f and Fig. S16 in Supporting information), including: (1) electrolysis for 2 h at open-circuit voltage; (2) electrolysis for 2 h at −0.1 V in Ar atmosphere; (3) electrolysis for 2 h at −0.1 V using carbon paper as the working electrode. It is found that NH3 is barely yielded in the control experiments, which confirms that NH3 is produced by electrocatalytic NRR of Au/ZrO2–800 catalyst.
This work synthesized nanomaterials with hetero-phase structures as NRR catalysts by simply changing the reaction temperature to pyrolyze MOF precusors. MOFs play a vital role in forming the mixed-component phases compared with other templates for synthesizing metal oxide. The obtained electrocatalyst Au/ZrO2–800 possessing roughly close proportions of monoclinic and tetragonal phases affords an excellent NRR performance with a high NH3 yield of 22.32 µg h−1 mgcat.−1 and a Faradaic efficiency of 31.92% at low overpotential. We believe this work will pave the way for exploring controllable preparation and precise synthesis of nano-hetero-phase structures.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation (Nos. 22075133 and 21701086).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108654.
[1]
W. Lin, H. Chen, G. Lin, et al., Angew. Chem. Int. Ed. 61 (2022) e202207807. doi: 10.1002/anie.202207807
[2]
S. Qiang, F. Wu, J. Yu, et al., Angew. Chem. Int. Ed. 62 (2023) e202217265. doi: 10.1002/anie.202217265
[3]
B.H.R. Suryanto, K. Matuszek, J. Choi, et al., Science 372 (2021) 1187–1191. doi: 10.1126/science.abg2371
T. Dambrauskas, H.H. Cornish, Am. Ind. Hyg. Assoc. J. 23 (1962) 151–156. doi: 10.1080/00028896209343218
Figure 1
(a) Scheme image of preparation of hetero-phase Au/ZrO2. (b) XRD patterns of hetero-phase Au/ZrO2. (c) TEM image of Au/ZrO2–800 composites. HRTEM images of (d) Au/ZrO2–700, (e) Au/ZrO2–800 and (f) Au/ZrO2–900. (g) STEM image and the corresponding EDX elemental mapping images of Au/ZrO2–800 composites.
Figure 3
Electrocatalytic NRR on Au/ZrO2–800 catalyst in 0.1 mol/L KOH electrolyte. (a) LSV curves in N2- and Ar-saturated electrolytes. (b) NH3 yield at different applied potentials. (c) FEs at diverse applied potentials. (d) Chronoamperometry test at –0.1 V for 24 h. (e) Comparison of electrocatalytic NRR on Au/ZrO2–700, Au/ZrO2–800 and Au/ZrO2–900 catalysts in 0.1 mol/L KOH electrolyte. (f) UV–vis absorption spectra under different conditions.

 DownLoad:
DownLoad:



 下载:
下载:




