Table 1.
The conversion of C4-C6 sugars toward its lactones.a
Citation:
Yulu Zhan, Yingshuang Hui, Shuqi Wang, Lou Gao, Yangbin Shen, Zhen-Hua Li, Yahong Zhang, Yi Tang. One-step selective dehydrogenation of cyclic hemiacetal sugars toward to their chiral lactones[J]. Chinese Chemical Letters,
2023, 34(4): 107677.
doi:
10.1016/j.cclet.2022.07.020

One-step selective dehydrogenation of cyclic hemiacetal sugars toward to their chiral lactones
English
One-step selective dehydrogenation of cyclic hemiacetal sugars toward to their chiral lactones
Abstract:
Chiral glycosyl lactone is an important class of bioactive compound and pharmaceutical intermediate in nature, especially for chiral lactones with 4 carbon atoms, which are very useful building blocks for synthesis of biologically interesting compounds. Herein, a selective dehydrogenation and solvent matched catalytic system under oxygen-free conditions was developed to try to achieve the one-step direct conversion of cyclic hemiacetal sugars toward their chiral glycosyl lactones. During the process, the inherent structural characteristics of sugar was efficiently utilized, and the transfer of its chiral centers was realized. Under the optimum condition, the corresponding lactones were successfully prepared from C4-C6 sugars with cyclic hemiacetal structure in acetonitrile. The reaction mechanism in acetonitrile was explored by the first principle density functional theory calculations and tracking reaction process. It was found that the high lactone yield in acetonitrile was due to the high proportion of α-conformation form among multiple tautomers in it. This selective dehydrogenation process may further extend the possibility of the preparation of chiral synthons from carbohydrates directly.
-
Key words:
- Biomass
- / Cyclic hemiacetal sugars
- / Dehydrogenation
- / Chiral lactones
- / Conformation
-
As the only readily renewable carbon‐based resource on earth, biomass can realize the sustainable production of liquid fuels and value-added chemicals. And it is a very important route to solve a series of problems caused by excessive exploitation of fossil resources, which has also been the major driving force for the evolution of modern chemical industry [1]. Natural carbohydrates with multi-functional groups are readily converted into flexible generic molecules for the production of renewable fuels and chemicals [2, 3]. Especially, their clearly-defined chirality centers and high abundance enable them to become ideal precursors in the design and synthesis of biologically natural products and bioactive molecules [4, 5].
Lactones are compounds formed by molecular lactonization of hydroxy fatty acid molecules and can be divided into different types depending on the position of the hydroxyl group. Glycosyl lactones are a widely occurring class of compounds with biological activity in nature, and are also useful intermediates in the synthesis of natural products. They are versatile food additives that are commonly used in the food industry as coagulants, stabilizers, sourdoughs, preservers, and preservatives [6, 7]. Currently, chiral glycosyl lactones are mostly obtained by enzymatic fermentation or chemical methods. Gluconolactone is usually synthesized by the oxidation of glucose with bromine followed by stepwise crystallization [8-10]. An alternative way to prepare glycosyl lactone is a transfer dehydrogenation reaction catalyzed by transition metals, which has been proved useful for protected and unprotected sugar alcohols as well as reducing sugars. Beaupère et al. used RuH2(PPH3)4 as catalyst and benzylidene acetone as the hydrogen acceptor to dehydrogenate various C4-C6 reducing sugars and unprotected sugar alcohols under mild conditions in DMF [11, 12], and nuclear magnetic resonance (NMR) characterization was employed to demonstrate their conformations [13, 14].
Among various glycosyl lactones, d-(-)-erythronolactone (ERL) containing two chiral centers has been reported that it can be converted into many chiral pharmaceutical intermediates such as 2,3-O-isopropylidene-D-erythronolactone and (2-azaallyl)stannane by its carbonyl addition and coupling reactions [15, 16], because chiral compounds with 3/4 carbon atoms are the optimum chiral synthon in size [17]. However, works on the preparation of C4 chiral synthons have rarely been published so far, especially for the synthesis of ERL. Traditionally, ERL could be obtained by the oxidation of aldehyde group in d-(-)-erythrose (ERO) and subsequent lactonization. During this process, the addition of strong oxidants such as hydrogen peroxide and strong acids such as sulfuric acid is necessary [18, 19]. Generally, ERO was produced by the oxidation of glucose in presence of lead(Ⅳ) tetraacetate and cobalt(Ⅱ) chloride [20]. Recently, Zhang's group has reported the conversion of d-hexoses toward ERO via a Lewis acid catalyzed retro-aldol process [21-23], laying a solid foundation for the conversion of ERO toward ERL. However, ERO is prone to dehydrate through anti-Michael reaction owing to the presence of carbonyl and β-hydroxy groups in it, which leads to its poor thermostability, especially in alkaline conditions [24]. This inherent property of ERO associated with its structure makes it difficult to selectively transform. Therefore, for the conversion of ERO toward its lactone, mild reaction conditions such as fewer process steps, shorter treatment time as well as no addition of oxidants or caustic alkali are more appropriate.
It is worthy to mention that catalytic acceptor-less dehydrogenation process, which provides a unique opportunity to release H2, is another convenient, atom-economical approach [25-27]. Obviously, glycosyl lactone is the direct dehydrogenation product of sugar with the cyclic hemiacetal structure. Moreover, the chirality of the corresponding lactone can be well retained because this oxidation is limited to anomeric hydroxyl group [28-30]. Inspired by this, herein, we proposed a one-step mild non-oxidative dehydrogenation route to directly convert cyclic hemiacetal sugars into chiral glycosyl lactones. As we expected, the chiral centers of sugar can be retained and transferred into lactone without prior protection or functionalization, meanwhile, H2 is observed at the same time. Solvents on the reaction process have been screened, and the highest yield of chiral glycosyl lactone has been obtained in acetonitrile (CH3CN). Experimental and theoretical evidences show that the highest ratio of α/β conformation of cyclic hemiacetal sugar in CH3CN is responsible for the high lactone yield in it. This research may provide some guidance for the generation of chiral lactones, thereby further enhancing the bioactivity and practical values of biomass in nature.
It is generally known that solvent has a notable influence on the reaction process, and the catalytic dehydrogenation reaction is also included [31-34]. Taking the flexible structure of sugar molecules in solvents into consideration, the effect of different solvents on the conversion of cyclic hemiacetal sugars toward lactones was explored firstly, and dimethyl sulfoxide (DMSO), N, N-dimethylformamide (DMF), CH3CN and methyl isobutyl ketone (MIBK) were adopted for comparison. Combined with our previous research [35], herein, [Cp*Ir(bpyO)]OH− (Cp* = 1,2,3,4,5-pentamethyl-cyclopentadienide, bpyO = α,α'-bipyridonate) was utilized to perform the acceptorless dehydrogenation process of C4-C6 cyclic hemiacetal sugars. The structure of [Cp*Ir(bpyO)]OH− catalyst is given in Fig. S1 (Supporting information). Table 1 shows the products distribution of the dehydrogenation process of C4-C6 sugars. It is interesting that for C4-C6 sugars, CH3CN is the reaction solvent with the highest yield of each glycosyl lactone and the highest carbon balance. Specifically, the yield of d-glucono-1,4-lactone from glucose in CH3CN is 35.9%, times more than that in DMF and MIBK. Besides lactone, isomeric products, C-C bond breaking products can also be observed in the reaction solution. For mannose, the yield of lactone in CH3CN is 32.4%, much higher than 12.9% in DMF and 8.3% in MIBK. The yield of d-glucono-1,4-lactone from fructose in CH3CN is 26.8%, which is 5~6 times higher than that in DMF and MIBK. Likewise, for xylose, it is much less converted in CH3CN than the other three solvents, but the d-xylono-1,4-lactone is produced in comparable yield. Obviously, CH3CN is very favour of the dehydrogenation process of cyclic hemiacetal sugar in the presence of [Cp*Ir(bpyO)]OH−, and so suppresses the other side reaction of sugars caused by its instable structure. Therefore, we assume that the phenomenon may be related to the existence of different conformational forms of cyclic hemiacetal sugars, which were also verified in subsequent results.
Table 1
More specific, it is worth mentioning that except catalyst, an alkali, i.e., NaOH, has to be added for the conversion of C5-C6 sugars. For example, glucose displays a relatively low conversion (20.1%) and the low yield of d-glucono-1,4-lactone (7.5%) in CH3CN when the addition of NaOH is absent. Only with the addition of NaOH in CH3CN (OH−/glucose molar ratio = 0.25), the conversion of glucose sharply rises to 85.5%, and the yield of d-glucono-1,4-lactone increases by four times from 7.5% to 35.9% (Table 1). This can be attributed that alkaline environment is conducive to the dehydrogenation process [36]. However, for ERO with 4 carbon atoms, without the addition of alkali, 41.7% yield of ERL can be obtained in CH3CN. Besides, gas chromatography is employed to determine the gaseous product, and H2 can be solely detected (Fig. S2 in Supporting information). Obviously, the catalytic acceptorless dehydrogenation process catalyzed by [Cp*Ir(bpyO)]OH− is more suitable for the conversion of ERO toward ERL.
Furthermore, the optimization of reaction condition and the detailed product characteristics of ERO conversion was studied. To achieve ERL efficiently, we dehydrogenated ERO in CH3CN within a certain temperature range. The results in Table 1 show that the ERO conversion increases constantly with the increase of reaction temperature. Interestingly, as the reaction temperature increases from 120 ℃ to 150 ℃, the yield of ERL significantly increases from 3.7% to 41.7%. However, as the temperature continues to increase to 180 ℃, the yield of ERL decreases to 30.1%. This is presumably due to the poor stabilities of ERO and ERL at high temperature. Different molar ratios of catalyst to ERO (0.01, 0.02 and 0.04) were employed to explore the influence of catalyst amount on dehydrogenation reaction (Fig. S3a in Supporting information). As we can see, both the conversion of ERO and the yield of ERL increase first and then decrease with the increasing catalyst amount. More specifically, when the catalyst/ERO molar ratio is lower than 0.02, the ERO conversion increases with the increasing molar ratio. However, as the molar ratio continues to rise to 0.04, the conversion of ERO declines from 62.4% to 38.3%. The yield of ERL follows a similar pattern, which sharply drops from 41.7% to 15.2%. The highest conversion of ERO (62.4%) and the highest yield of ERL (41.7%) are obtained at the catalyst/ERO molar ratio of 0.02. The fast decreasing of the ERL yield at a high catalyst/ERO molar ratio can be assigned to the reversible hydrogenation ability of [Cp*Ir(bpyO)]OH− [37, 38]. Meanwhile, as Fig. S3b (Supporting information) shows, as the ERO concentration increases from 0.04 mol/L to 0.08 mol/L, the yield of ERL dramatically increases from 18.3% to 41.7%. Further increasing the ERO concentration to 0.16 mol/L, the yield of ERL decreases rapidly to 10.6%. Finally, optimum conditions, i.e., molar ratio of catalyst/sugar = 0.02, sugar of 0.08 mol/L as well as the reaction temperature of 150 ℃ in CH3CN, are adopted for the further study.
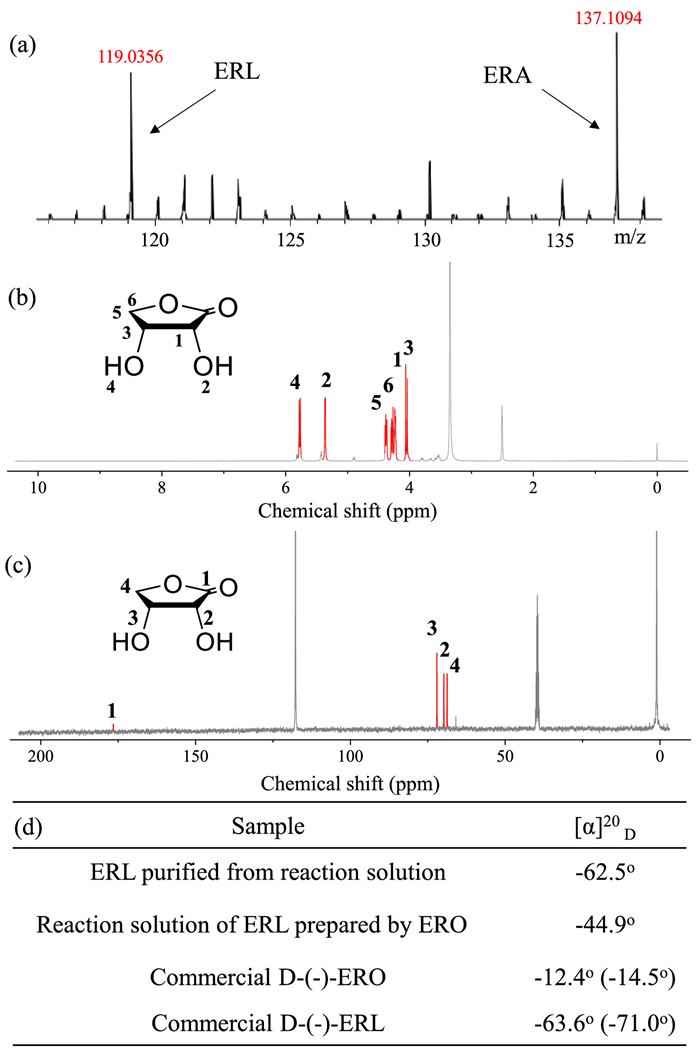
The formation and chirality of ERL were confirmed by mass spectrometry (MS), 1H NMR, 13C NMR and specific rotation (Fig. 1). Two signals at m/z 119 and 137 in the positive ionization mode corresponding to the theoretical m/z values of ERL and d-(-)-erythronic acid (ERA) (Fig. 1a), respectively. Peaks with the same m/z values are clearly observed from the crude product mixture as well as the commercial ERL solution (Fig. S4 in Supporting information). It should be noted that the appearance of ERA signal is mainly due to the hydrolysis of ERL at 180 ℃ during MS detection. Furthermore, the resonance signals obtained from its 1H NMR (δ: CH 4.01, 4.05, CH2 4.25, 4.38, OH 5.36, 5.78) and 13C NMR (δ: C=O 178.2, CH2 63.8, CH 69.0, 71.5, 73.7, 76.7) spectra in Figs. 1b and c are basically coincident with those of commercial ERL (Fig. S5 in Supporting information). Moreover, the specific rotation (−62.5°) of the ERL purified from the reaction is very close to that of the commercial ERL (−63.6°) as well as theoretical value for this compound. The discrepancy with the theoretical value mainly originates from the influence of different solvents. Besides, the change in the specific rotation from −12.4° to −44.9° after the reaction reveals the formation of levorotatory products with higher left-handed values from ERO (Fig. 1d), which further demonstrates the successful preparation of ERL and the retainment of its natural chirality.
Figure 1
 Figure 1. (a) Mass spectrometry detection of the purified ERL solution in the positive ionization mode. (b) 1H NMR and (c) 13C NMR spectra of the purified ERL solution (~120 ppm was assigned to the resonance signal of CN in CH3CN), and DMSO-d6 was used as the field-locking reagent. (d) Specific rotations of the purified ERL, reaction solution and commercial d-(-)-ERO and d-(-)-ERL in CH3CN. The concentration of all the aqueous solutions was fixed at 1.0 wt%. The data in parentheses represent their theoretical values in water.
Figure 1. (a) Mass spectrometry detection of the purified ERL solution in the positive ionization mode. (b) 1H NMR and (c) 13C NMR spectra of the purified ERL solution (~120 ppm was assigned to the resonance signal of CN in CH3CN), and DMSO-d6 was used as the field-locking reagent. (d) Specific rotations of the purified ERL, reaction solution and commercial d-(-)-ERO and d-(-)-ERL in CH3CN. The concentration of all the aqueous solutions was fixed at 1.0 wt%. The data in parentheses represent their theoretical values in water.Likewise, for C5/6 at room temperature, water was adopted as the solvent for measuring the specific rotation. Concretely, the change in the specific rotation from +52.5° to +20.3° after the glucose reaction indicates the generation of levorotatory products or massive dextrorotatory products with lower right-handed values (d-gluconic acid) (Table S1 in Supporting information). For fructose reaction, the change in the specific rotation from −91.9° to −36.5° demonstrates that the dextrorotatory products (d-glucono-1,4-lactone) obtained. For mannose reaction, the specific rotation varied from +13.8° to +18.9°, which proves that substances with a higher right-handed value (d-mannono-1,4-lactone) is generated. Similarly, the formation of d-xylono-1,4-lactone can be obviously inferred from the change of the specific rotation of xylose reaction solution. These results further confirm that this selective dehydrogenation process catalyzes the oxidation of anomeric hydroxyl group rather than other hydroxyl group. Otherwise, the specific rotation of the solution will change randomly until racemization. In addition, the mass spectrometry analysis was utilized to detect the glycosyl lactones derived from C5/6 sugars in CH3CN. As shown in Fig. S6 (Supporting information), the corresponding glycosyl lactones were clearly visible in the negative ionization mode.
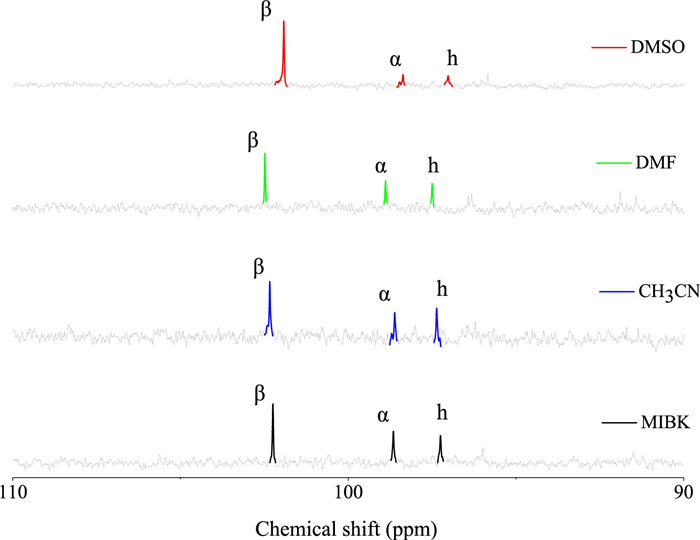
For substance with multiple tautomers, the reaction rate is dominated by the concentration of one or more tautomers rather than the total concentration of substrate if their interconversion is slow [39, 40]. As mentioned above, these cyclic hemiacetal sugars show various dehydrogenation activities in different organic solvents, and the highest yields of glycosyl lactones were obtained in CH3CN. Consequently, the complicated tautomeric composition in different solvents due to its structural diversity should be clarified to understand the effect of solvent. Previous 1H and 13C NMR measurements on ERO in aqueous solution revealed the co-existence of α- and β-furanose forms (90%) and they are in equilibrium with a considerable amount (~12%) of acyclic forms [41, 42]. Herein, 1H NMR and 13C NMR spectroscopies were employed to evaluate the tautomeric composition of ERO in the above several organic solvents. To better control variables, concentrations of ERO in different organic solvents are consistent. As shown in Fig. S7 (Supporting information) and Fig. 2, the chemical shifts (δH and δC) of ERO in different solvents are not identical. For 1H NMR spectra (Fig. S7), resonances at ~9.7 ppm are assigned to aldehyde H, δH between 4.8~5.6 ppm belong to α- and β-furanoses and linear hydrate (h) of ERO [41]. In their 13C NMR spectra (Fig. 2), α- and β-furanoses and h of ERO resonate between 95~110 ppm. For accurate quantitation, peak areas are determined by integration after amplification. For example, taking CH3CN solution as an example, for 1H NMR spectra in Fig. S7, a doublet of doublets at 5.36 ppm is assigned to β-furanose, and α-furanose is also a doublet of doublets at 5.38 ppm. In 13C NMR spectra (Fig. 2), a single of singlet at ~97 ppm is assigned to h. And single of singlet at ~102 ppm and ~99 ppm are assigned to α-furanose and β-furanose, respectively [43, 44]. Table S2 (Supporting information) shows the proportion of different conformations in selected solvents, which is determined by the peak intensities of different conformations in 13CNMR spectra (Fig. 2). As we can see, the peak intensity ratio for α/β increases in the following order: DMSO < DMF < MIBK < CH3CN. This trend correlates well with the trend in the yields of ERL in them, that is, the proportion of α-furanoses in CH3CN is the largest among these solvents, and the yield of ERL in it is the highest. To confirm this, the conversion of ERO in water was carried out. Because ERO in water possesses the same α/β ratio with DMF (Table S2, entries 2 and 5), the same yield of ERL should be observed. As shown in Table 1, the ERL yield in water is between those in DMSO and MIBK, but slightly lower than that in DMF. This can be attributed to the hydrolysis of ERL toward ERA in water (Fig. S8 in Supporting information). Obviously, the total yield of ERL and ERA in water is comparable with that in DMF. Consequently, it is expected that the content of α-furanoses in solution is directly proportional to the dehydrogenation rate of catalyst. This correlation between the α-furanose form and the lactone yield demonstrates again that this selective dehydrogenation process occurs on anomeric carbon atom of sugar.
Figure 2
 Figure 2. 13C NMR spectra of the ERO in DMSO, DMF, CH3CN and MIBK. DMSO-d6 was used as the field-locking reagent.
Figure 2. 13C NMR spectra of the ERO in DMSO, DMF, CH3CN and MIBK. DMSO-d6 was used as the field-locking reagent.Moreover, the existence forms of C5/6 cyclic hemiacetal sugars have been extensively studied by NMR spectroscopy [45, 46]. As shown in Fig. S9 (Supporting information), 13C NMR results show the conformational compositions of C5/6 sugars in CH3CN, and the assignment of peaks is done based on literatures [47-50]. Table S3 (Supporting information) shows the relative conformation proportions of C5/6 sugars calculated from 13C NMR spectra. The α-pyranose was found to account for 47% and the β-pyranose for 53% of the total glucose. For fructose, the α-furanose proportion is only 34%, while, the sum of β-furanose and β-pyranose is 66%. For mannose and xylose, they show the same ratio of α/β. The above results successfully proved the existence of α-conformation in CH3CN, and thus further proved our previous speculation, that is, a high α/β ratio leads to a high selectivity of lactone.
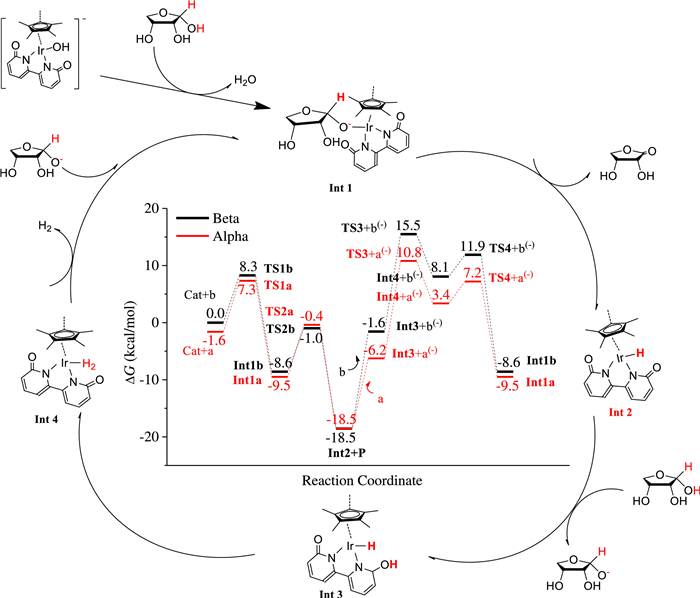
DFT calculations were performed to study the reaction mechanism of ERO to ERL catalysed by [Cp*Ir(bpyO)]OH−. The reaction pathway and its Gibbs free-energy profile at 298.15 K are presented in Scheme 1. In the Gibbs free-energy profile scheme, the red lines are for α-ERO and the black ones are for β-ERO, and names for the species ending with letter "a/b" are for α-/β-ERO, correspondingly. In the first step, the hydroxyl ion of [Cp*Ir(bpyO)]OH− abstracts one proton from α-/β-ERO leaving as water and the resulting ERO ion binds with Ir to form Int1a/Int1b in one elementary reaction step. Subsequently, Ir of Int1a/Int1b abstracts another proton from ERO ion to generate ERL (P) and Int2. Then, Int2 further abstracts one proton from another α-/β-ERO molecule using the oxygen atom of its C=O group, producing Int3 and ERO ion a(-)/b(-). The two hydrogen atoms on Int3, one bonding with Ir and the other bonding with O of C=O, form chemically adsorbed H2 on Ir, i.e. Int4. In the next step, H2 leaves from Ir and then ERO ion a(-)/b(-) binds immediately to Ir to form Int1a/Int1b and completes the catalytic cycle. From Scheme 1, it can be concluded that [Cp*Ir(bpyO)]OH− is just a pre-catalyst. The real catalyst is Int2 because it is the most stable intermediate in the reaction pathway and its generation is fast because the free-energy barriers of the reaction steps leading to it are all below 10 kcal/mol. The rate of the whole catalytic reaction is determined by two consecutive steps. The first step is from Int2 + ERL to Int3 + a(-)/b(-) and the second step is from Int3 to Int4. The overall free-energy barrier of this catalytic reaction is the free-energy difference between the highest transition state (TS3 + a(-)/b(-)) and the lowest intermediate (Int2 + P) in the catalytic cycle, which is 29.3/34.0 kcal/mol for the α-/β-ERO, respectively. The lower free- energy barrier for α-ERO is a result of a(-) being more stable than b(-), which leads to a lower free-energy change from Int2 + ERL to Int3 + a(-). The reason that a(-) is more stable than b(-) is that the oxyl ion in a(-) is stabilized by two adjacent OH groups through intra-hydrogen bonds while in b(-1) there are no OH groups to stabilize the oxyl ion because the OH groups and the oxyl ion are at the opposite side of the ring.
Scheme 1
 Scheme 1. Proposed reaction mechanism of dehydrogenation process of α-ERO based on DFT calculations, the critical reaction intermediate is Int2, and the active hydrogen atoms of ERO are highlighted in red. In the center, the DFT computed free-energy profile of the ERO dehydrogenation reaction is displayed.
Scheme 1. Proposed reaction mechanism of dehydrogenation process of α-ERO based on DFT calculations, the critical reaction intermediate is Int2, and the active hydrogen atoms of ERO are highlighted in red. In the center, the DFT computed free-energy profile of the ERO dehydrogenation reaction is displayed.The computational results show that the conversion of α-ERO to ERL has a lower overall free-energy barrier than that of β-ERO. Therefore, if the conversion among various conformers of ERO is slower than the dehydrogenation reaction, we would expect that the higher proportion of α-ERO is in the reaction system, the higher the yield of ERL would be. A recent theoretical study showed that with the help of water molecules, the conversion of α-ERO to other tautomers will still overcome a free-energy barrier of 40.7 kcal/mol [51]. This is too high a barrier to overcome under the present reaction temperature. It is expected that the conversion between α- and β-ERO would be even slower in anhydrous system without the help of water molecules. This clearly explains why the highest yield of ERL is obtained in CH3CN, because the proportion of α-ERO is the highest in it (Fig. 2 and Table S2).
To verify the theoretical prediction that Int2 is the working catalyst, we recorded the 1H NMR spectra of the reaction system under an excessive amount of [Cp*Ir(bpyO)]OH− in the reaction solution (Fig. S10 in Supporting information). A peak at −13.8 ppm appears after reaction for 25 min (Fig. S10b), which is assigned to the hydride H on Ir (Int2) [52]. As the reaction progresses to maximum yield, the active intermediate is difficult to detect due to its small amount (Fig. S10c). However, as exhibited in the Fig. S11 (Supporting information), after reaction, a typical Ir-H stretching at 2033 cm−1 was identified performing diffuse reflectance infrared spectroscopy (DRIFTS) on the spent catalyst. This is close to the position of Ir-H infrared vibration peak in literature, it indicates the removed H tends to adsorb on the iridium atom of the [Cp*Ir(bpyO)]OH−, which could lead to the structure change of the iridium complexes. This result well matches the theoretical prediction, that is, Int2 is the most stable intermediate and its generation is fast.
To sum up, we developed a mild one-step catalytic system without additional oxidant to convert cyclic hemiacetal sugar toward glycosyl lactones with its chirality delivery. Under the optimum conditions, high yield of lactone is obtained from various sugars in CH3CN. Moreover, up to 41.7% yield of ERL is obtained with a ERO conversion of 62.4% in CH3CN with no additives are required. An in-depth discussion and study of the product structure is provided by means of specific rotation test, NMR, FTIR and mass spectrometry. In addition, DFT calculations confirm that the tautomeric composition in solvent is the pivotal factor for this efficient conversion, and the high lactone yield in CH3CN can be assigned to the high proportion of α-conformation form of cyclic hemiacetal sugar in it. These findings not only provide a new insight into the retainment of chiral centre but also open up a promising route for the preparation of chiral synthons from biomass sugars in the future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Major Research and Development Plan (No. 2018YFA0209402), the National Natural Science Foundation of China (Nos. 21673045, 22088101).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:
10.1016/j.cclet.2022.07.020 .
-
-
[1]
J.P. Lange, ChemSusChem 11 (2018) 997-1014. doi: 10.1002/cssc.201702362
-
[2]
Z.J. Witczak, Curr. Med. Chem. 6 (1999) 165-178. doi: 10.2174/0929867306666220207213740
-
[3]
L.V. R. Reddy, V. Kumar, R. Sagar, et al., Chem. Rev. 113 (2013) 3605-3631. doi: 10.1021/cr200016m
-
[4]
C.H. Zhou, X. Xia, C.X. Lin, et al., Chem. Soc. Rev. 40 (2011) 5588-5617. doi: 10.1039/c1cs15124j
-
[5]
H.V. Scheller, P. Ulvskov, Annu. Rev. Plant Biol. 61 (2010) 263-289. doi: 10.1146/annurev-arplant-042809-112315
-
[6]
W. Terra, I. Terra, C. Ferreira, Int. J. Biochem. 15 (1983) 143-146. doi: 10.1016/0020-711X(83)90056-3
-
[7]
Y.B. Shen, Y.L. Zhan, S.P. Li, et al., ChemSusChem 11 (2018) 864-871. doi: 10.1002/cssc.201702359
-
[8]
E. Loubaki, S. Sicsic, F. Legoffic, Eur. Polym. J. 25 (1989) 379-384. doi: 10.1016/0014-3057(89)90154-7
-
[9]
M. Bucko, P. Gemeiner, A. Vikartovská, et al., Artif. Cells Blood Substit. Immobil. Biotechnol. 38 (2010) 90-98. doi: 10.3109/10731191003634745
-
[10]
H. Isbell, C. Hudson, Bur. Standards J. Res. 8 (1932) 327-338. doi: 10.6028/jres.008.025
-
[11]
M. Saburi, Y. Ishii, N. Kaji, et al., Chem. Lett. 18 (1989) 563-566. doi: 10.1246/cl.1989.563
-
[12]
Y.B. Shen, C. Bai, Y.L. Zhan, et al., ChemPlusChem 85 (2020) 1646-1654. doi: 10.1002/cplu.202000394
-
[13]
I. Isaac, I. Staslk, D. Beaupère, et al., Tetrahedron Lett. 36 (1995) 383-386. doi: 10.1016/0040-4039(94)02262-A
-
[14]
M.J. Mays, M.J. Morris, P.R. Raithby, et al., Organometallics 8 (1989) 1162-1167. doi: 10.1021/om00107a006
-
[15]
L.L. Wong, R.L. Wong, G. Loh, et al., Org. Process Res. Dev. 16 (2012) 1003-1012. doi: 10.1021/op200352k
-
[16]
Y. Novikov, S.D. Copley, B.E. Eaton, Tetrahedron Lett. 52 (2011) 1913-1915. doi: 10.1016/j.tetlet.2011.02.045
-
[17]
W.Z. Zhang, J.C.K. Chu, K.M. Oberg, et al., J. Am. Chem. Soc. 137 (2015) 553-555. doi: 10.1021/ja510348p
-
[18]
A. Gypser, M. Flasche, H.D. Scharf, Liebigs Ann. Chem. 1994 (1994) 775-780. doi: 10.1002/jlac.199419940803
-
[19]
W. Overend, M. Stacey, L. Wiggins, J. Chem. Soc. 1 (1949) 1358-1363.
-
[20]
B.M. Kabyemela, T. Adschiri, R. Malaluan, et al., Ind. Eng. Chem. Res. 36 (1997) 2025-2030. doi: 10.1021/ie960747r
-
[21]
W. R Hou, Y. E Yan, G. Li, et al., ChemCatChem 11 (2019) 4182-4188. doi: 10.1002/cctc.201900094
-
[22]
Y. E Yan, L. Feng, G. Li, et al., ACS Catal. 7 (2017) 4473-4478. doi: 10.1021/acscatal.7b01159
-
[23]
S.Y. Lin, X. Guo, K. Qin, et al., ChemCatChem 9 (2017) 4179-4184. doi: 10.1002/cctc.201700944
-
[24]
M. Dusselier, P. Van Wouwe, S. De Smet, et al., ACS Catal. 3 (2013) 1786-1800. doi: 10.1021/cs400298n
-
[25]
R. Kawahara, K. i. Fujita, R. Yamaguchi, J. Am. Chem. Soc. 134 (2012) 3643-3646. doi: 10.1021/ja210857z
-
[26]
N. Sieffert, M. Buehl, J. Am. Chem. Soc. 132 (2010) 8056-8070. doi: 10.1021/ja101044c
-
[27]
M. Kamitani, M. Ito, M. Itazaki, et al., Chem. Commun. 50 (2014) 7941-7944. doi: 10.1039/C4CC01498G
-
[28]
J. Zhang, G. Leitus, Y. Ben-David, et al., J. Am. Chem. Soc. 127 (2005) 10840-10841. doi: 10.1021/ja052862b
-
[29]
S. Chakraborty, P.O. Lagaditis, M. Foerster, et al., ACS Catal. 4 (2014) 3994-4003. doi: 10.1021/cs5009656
-
[30]
M. Ohno, M. Otsuka, Org. React. 37 (2004) 1-55.
-
[31]
L. Moity, V. Molinier, A. Benazzouz, et al., Green Chem. 18 (2016) 3239-3249. doi: 10.1039/C6GC00112B
-
[32]
A.S. Amarasekara, L.D. Williams, C.C. Ebede, Carbohydr. Res. 343 (2008) 3021-3024. doi: 10.1016/j.carres.2008.09.008
-
[33]
J.L. Santos, C. Megías-Sayago, S. Ivanova, et al., Chem. Eng. J. 420 (2021) 127641. doi: 10.1016/j.cej.2020.127641
-
[34]
Q.Y. Li, S.Y. Cheng, H.T. Tang, et al., Green Chem. 21 (2019) 5517-5520. doi: 10.1039/c9gc03028j
-
[35]
Y.L. Zhan, W.R. Hou, G. Li, et al., ACS Sustain. Chem. Eng. 7 (2019) 17559-17564. doi: 10.1021/acssuschemeng.9b04617
-
[36]
K. Fujita, R. Kawahara, T. Aikawa, et al., Angew. Chem. Int. Ed. 54 (2015) 9057-9060. doi: 10.1002/anie.201502194
-
[37]
R. Yamaguchi, C. Ikeda, Y. Takahashi, et al., J. Am. Chem. Soc. 131 (2009) 8410-8412. doi: 10.1021/ja9022623
-
[38]
R. Kawahara, K. i. Fujita, R. Yamaguchi, Angew. Chem. Int. Ed. 51 (2012) 12790-12794. doi: 10.1002/anie.201206987
-
[39]
S.J. Angyal, Angew. Chem. Int. Ed. 8 (1969) 157-166. doi: 10.1002/anie.196901571
-
[40]
S.J. Angyal, V.A. Pickles, Aust. J. Chem. 25 (1972) 1695-1710. doi: 10.1071/CH9721695
-
[41]
A.S. Serianni, J. Pierce, S.G. Huang, et al., J. Am. Chem. Soc. 104 (1982) 4037-4044. doi: 10.1021/ja00379a001
-
[42]
J.M. Risley, R.L. Van Etten, Biochemistry 21 (1982) 6360-6365. doi: 10.1021/bi00268a007
-
[43]
A.S. Serianni, J. Pierce, R. Barker, Biochemistry 18 (1979) 1192-1199. doi: 10.1021/bi00574a012
-
[44]
A.S. Serianni, E.L. Clark, R. Barker, Carbohydr. Res. 72 (1979) 79-91. doi: 10.1016/S0008-6215(00)83925-7
-
[45]
M. Maebayashi, M. Ohba and T. Takeuchi, J. Mol. Liq. 232 (2017), 408-415. doi: 10.1016/j.molliq.2017.02.062
-
[46]
A. Perlin, B. Casu, H. Koch, Can. J. Chem. 48 (1970) 2596-2606. doi: 10.1139/v70-435
-
[47]
S.R. Maple, A. Allerhand, J. Am. Chem. Soc. 109 (1987) 3168-3169. doi: 10.1021/ja00244a063
-
[48]
E. Alexandersson, G. Nestor, Carbohydr. Res. 511 (2022) 108477. doi: 10.1016/j.carres.2021.108477
-
[49]
P. Qrtiz, E. Reguera, J. Fernández-Bertrán, J. Fluor. Chem. 113 (2002) 7-12. doi: 10.1016/S0022-1139(01)00399-2
-
[50]
M.J. King-Morris, A.S. Serianni, J. Am. Chem. Soc. 109 (1987) 3501-3508. doi: 10.1021/ja00246a001
-
[51]
M. Szczepaniak, J. Moc, J. Comput. Chem. 38 (2017) 288-303. doi: 10.1002/jcc.24680
-
[52]
A.H. Ngo, M. Ibañez, L.H. Do, ACS Catal. 6 (2016) 2637-2641. doi: 10.1021/acscatal.6b00395
-
[1]
-
Figure 1 (a) Mass spectrometry detection of the purified ERL solution in the positive ionization mode. (b) 1H NMR and (c) 13C NMR spectra of the purified ERL solution (~120 ppm was assigned to the resonance signal of CN in CH3CN), and DMSO-d6 was used as the field-locking reagent. (d) Specific rotations of the purified ERL, reaction solution and commercial d-(-)-ERO and d-(-)-ERL in CH3CN. The concentration of all the aqueous solutions was fixed at 1.0 wt%. The data in parentheses represent their theoretical values in water.
Figure 2 13C NMR spectra of the ERO in DMSO, DMF, CH3CN and MIBK. DMSO-d6 was used as the field-locking reagent.
Scheme 1 Proposed reaction mechanism of dehydrogenation process of α-ERO based on DFT calculations, the critical reaction intermediate is Int2, and the active hydrogen atoms of ERO are highlighted in red. In the center, the DFT computed free-energy profile of the ERO dehydrogenation reaction is displayed.
-

 DownLoad:
DownLoad:


 下载:
下载:


 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 789
- HTML全文浏览量: 111
