引用本文:
郭征楠, 刘峥, 魏席, 唐群, 黎焕林. 以1,2,4,5-苯四甲酸或1,2,3,4-丁烷四羧酸为配体的金属有机框架的合成、表征及性质[J]. 无机化学学报,
2016, 32(1): 9-17.
doi:
10.11862/CJIC.2016.028

Citation: GUO Zheng-Nan, LIU Zheng, WEI Xi, TANG Qun, LI Huan-Lin. Syntheses, Characterization and Properties of Metal-Organic Frameworks based on 1,2,4,5-Benzene Tetracarboxylic Acid or 1,2,4,5-Butane Tetracarboxylic Acid[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(1): 9-17. doi: 10.11862/CJIC.2016.028
Citation: GUO Zheng-Nan, LIU Zheng, WEI Xi, TANG Qun, LI Huan-Lin. Syntheses, Characterization and Properties of Metal-Organic Frameworks based on 1,2,4,5-Benzene Tetracarboxylic Acid or 1,2,4,5-Butane Tetracarboxylic Acid[J]. Chinese Journal of Inorganic Chemistry, 2016, 32(1): 9-17. doi: 10.11862/CJIC.2016.028
以1,2,4,5-苯四甲酸或1,2,3,4-丁烷四羧酸为配体的金属有机框架的合成、表征及性质
摘要:
以1,2,4,5-苯四甲酸(H4BETA)与1,2,3,4-丁烷四羧酸(H4BTCA)为有机配体,采用溶剂热法,成功合成了3个金属有机框架(Metal-Organic Frameworks, MOFs):{[Co5(BETA)2(OH)2(H2O)2]·10H2O}n(1)、{[Zn5(BETA)2(OH)2(H2O)2]·10H2O}n(2)和{[Mn2(BTCA)(H2O)3]·H2O}n(3),并利用X射线单晶衍射、红外光谱(IR)、荧光光谱(PL)和热重分析(TG/DTG)等测试手段对其进行了表征。单晶结构研究表明3个MOFs均属于单斜晶系,P21/n空间群;框架1和2都是由1,2,4,5-苯四甲酸与五核的金属簇连接形成三维框架结构,沿a轴方向具有一维孔道结构,孔径大小为0.990 nm×1.307 nm;框架3是由1,2,3,4-丁烷四羧酸与Mn2+连接形成4,8-c网络的三维框架结构。
English
Syntheses, Characterization and Properties of Metal-Organic Frameworks based on 1,2,4,5-Benzene Tetracarboxylic Acid or 1,2,4,5-Butane Tetracarboxylic Acid
Abstract:
Three Metal-Organic Frameworks (MOFs) materials, namely, {[Co5(BETA)2(OH)2(H2O)2]·10H2O}n(1), {[Zn5(BETA)2(OH)2(H2O)2]·10H2O}n(2) and {[Mn2(BTCA)(H2O)3]·H2O}n (3), (H4BETA=1,2,4,5-benzene tetracarboxylic acid; H4BTCA=1,2,4,5-butane tetracarboxylic acid), have been solvothermally synthesized, and characterized by X-ray single crystal diffraction, IR spectrum, PL spectrum, and TG/DTG analyses. The results show that the three MOFs belong to monoclinic system, space group P21/n. Five-nuclear metal clusters in compound 1 and 2 are linked by BETA4- ligand to form 3D framework structure, and produce 1D channel with the pore size of 0.990 nm×1.307 nm along the a axis. The framework of compound 3 is consisted of Mn2+ ions with BTCA4- ligands forming 4,8-c network. CCDC: 1409602, 3; 1409603, 2; 1409601, 3.
-
Key words:
- MOFs
- / carboxylate ligand
- / synthesis
- / property
-
金属-有机框架(Metal-Organic Frameworks, MOFs)[1]是一类由有机配体和无机金属单元通过共价键和(或)氢键等形成的从一维到三维的无限网状结构[2-3]。目前,已经有大量的MOFs材料被合成,主要是以含羧基有机配体为主,或与含氮杂环有机中性配体共同使用。这些MOFs材料中多数都具有高的孔隙率和较大的比表面积[4-7]。因此,在气体吸附、生物医学、光电材料、催化剂[8-10]等方面具有巨大的应用潜力。1999年,美国的Yaghi教授以对苯二甲酸(1.4-H2BDC)为配体,合成了MOF-5[11]。在随后的几年里他又对MOF-5进行了修饰和加工,进一步合成了新一代IRMOF(Isoreticular Metal-Organic Framework)[12],在孔径和稳定性方面取得了极大的改善。
MOFs材料的应用研究领域十分广阔,应用前景十分诱人。而对于MOFs的制备,常采用室温挥发法、溶剂(水)热法、扩散法 [13-16]等方法。由于1, 2, 4, 5-苯四甲酸(H4BETA)的苯环与羧基在同一平面上,可作为理想的四边形构型连接点,较易形成多孔道MOFs,此外,通过仔细控制反应条件,部分或完全发生去质子化,更加容易形成高核MOFs[17];而1, 2, 3, 4-丁烷四羧酸(H4BTCA)的烷链多羧酸框架可任意旋转来构筑多孔MOFs[18],比起其他类型的多羧酸配体更加有吸引力和挑战性。
本文采用H4BETA和H4BTCA为配体,Co、Zn、Mn为中心离子,利用水热法合成了3个MOFs材料,并获得了单晶。通过X射线单晶衍射、元素分析、红外光谱(IR)、荧光光谱(PL)和热重分析(TG/DTG)等技术对合成的MOFs材料的晶体结构、谱学性质和热稳定性进行表征。
1 实验部分
1.1 仪器与试剂
日本Hitch公司F-4600荧光分光光谱仪;美国Perkin-Elmer 2400 CHN元素分析仪;日本岛津公司Shimadzu FTIR-8400红外光谱仪;德国耐驰公司STA-449热分析仪;德国Bruker公司Bruker SMART APEX CCD单晶衍射仪。六水合硝酸钴、氯化锰、硝酸锌、H4BETA、H4BTCA均为分析纯试剂。
1.2 合成
1.3 晶体结构测定
选取尺寸分别为0.15 mm×0.10 mm×0.09 mm (1)、0.17 mm×0.14 mm×0.11 mm (2)和0.21 mm×0.20 mm×0.19 mm (3)的晶体,用Bruker SMART APEX CCD单晶衍射仪依次收集数据。采用经石墨单色化的Mo Kα射线(λ=0.071 073 nm),用φ-ω扫描模式分别在5.96°~58.22°、5.7°~58.26°、6.0°~50.18°(2θ) 范围内于293(2) K收集衍射点,衍射强度数据用SADABS程序进行经验吸收校正[19]。结构用SHELXS-97程序通过直接法解出[20],对非氢原子及其各向异性温度因子用 SHELXL-97程序进行全矩阵最小二乘法修正[21]。所有氢原子均为理论加氢。有关晶体学和结构修正数据见表 1。
 表 1
化合物1、2、3的晶体学数据
Table 1.
Crystal data of compound 1, 2 and 3
表 1
化合物1、2、3的晶体学数据
Table 1.
Crystal data of compound 1, 2 and 3

CCDC:1409602,1; 1409603,2;1409601,3。
1.2.2 {[Zn5(BETA)2(OH)2(H2O)2]·10H2O}n(2)的合成
2的合成与1类似,只是将Co(NO3)2·6H2O换为Zn(NO3)2·6H2O。无色针状晶体,产率75%。元素分析按Zn5C20O30H30计算的理论值(%):C, 22.29; H, 2.81; 实验值(%):C, 22.16; H, 2.87。
1.2.3 {[Mn2(BTCA)(H2O)3]·H2O}n(3)的合成
3的合成与1类似,只是将Co(NO3)2·6H2O换为MnCl2·4H2O,将配体换为H4BTCA。无色块状晶体,产率68%。元素分析按Mn2C8O12H10计算的理论值(%):C, 23.55; H, 2.47;实验值(%):C, 23.64; H, 2.53。
1.2.1 {[Co5(BETA)2(OH)2(H2O)2]·10H2O}n(1)的合成
将含有0.2 mmol Co(NO3)2·6H2O、0.2 mmol H4BETA的15 mL水溶液在室温下磁力搅拌30 min,并用氨水调节pH值至7~8之间,继续搅拌30 min,然后将烧杯中的混合液转移到25 mL带有聚四氟乙烯衬底的水热反应釜中,加热到140 ℃晶化72 h。程序降温至100 ℃,保温10 h,自然冷却至室温,有粉色针状晶体生成,过滤,自然晾干后收集样品。产率71%。元素分析按Co5C20O30H30计算的理论值(%):C, 22.98; H, 2.89;实验值(%):C, 22.91; H, 2.82。
2 结果与讨论
2.1 晶体结构描述
晶体结构分析表明化合物1和2同构,属于单斜晶系,P21/n空间群,这里以化合物1为例来描述晶体结构。化合物1的部分键长键角见表 2、表 3。图 1为化合物1的晶体结构图,图 2为配位多面体图,图 3为化合物1的拓扑结构图。由图 1和图 2可见,每1个钴离子与6个氧结合,钴离子配位数为6,并通过三桥联的氧原子连接形成八面体对称空间构型的五核钴簇。5个钴离子有3种不同的配位环境(如图 2),中心离子Co1的配位环境中,O1和O1A来自水中的氧,O5、O5A和O11、O11A来自BETA4-上的羧基氧;在Co2的配位环境中,O1、O6来自于水中的氧,O2、O7、O8、O9B来自于BETA4-的羧基氧;Co3的配位环境中,O1、O10来自于羟基中的氧或配位水,O3、O4、O9、O11来自于BETA4-上的羧基氧。由表 1可知,Co-O的配位键长中最长为0.234 8 nm,最短为0.202 6 nm,与文献报道的含钴-羧基单元的化合物键长相符[22]。由图 3可见,1以五核钴簇为金属节点,以BETA4- 为连接器,采用四连接方式,形成1, 6-c网络,其拓扑符号为{44; 62}。化合物1沿a轴方向具有1D孔道,孔径大小为0.990 nm×1.307 nm,里面填充大量结晶水分子。
表 2
化合物1的主要键长(nm)和键角(°)
Table 2.
Selected bond lengths(nm) and bond angles(°) for compound 1
 表 3
化合物3的主要键长(nm)和键角(°)
Table 3.
Selected bond lengths(nm) and bond angles (°) for compound 3
表 3
化合物3的主要键长(nm)和键角(°)
Table 3.
Selected bond lengths(nm) and bond angles (°) for compound 3

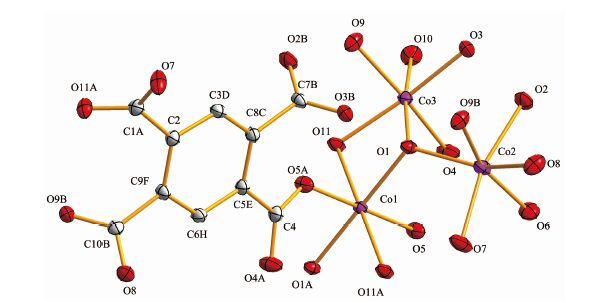 图 1
化合物1的晶体结构图(50%椭球概率)
Figure 1.
Crystal structure of the compound 1 shown as 50% thermal ellipsoid probability
图 1
化合物1的晶体结构图(50%椭球概率)
Figure 1.
Crystal structure of the compound 1 shown as 50% thermal ellipsoid probability
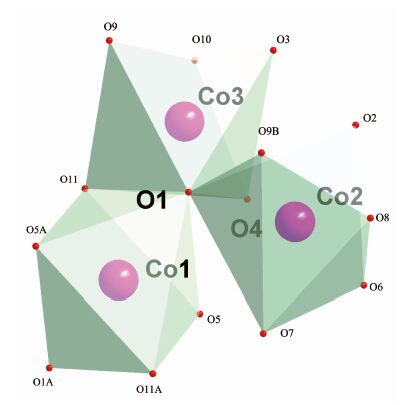 图 2
化合物1中CoⅡ的配位多面体图
Figure 2.
Coordination polyhedron for CoⅡ in 1
图 2
化合物1中CoⅡ的配位多面体图
Figure 2.
Coordination polyhedron for CoⅡ in 1
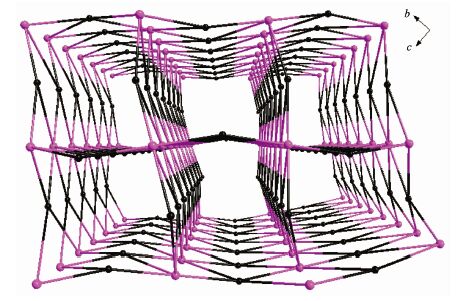 图 3
化合物1和2的拓扑结构图
Figure 3.
Topological net of compound 1 and 2
图 3
化合物1和2的拓扑结构图
Figure 3.
Topological net of compound 1 and 2
化合物3的部分键长键角见表 3。图 4为化合物3的晶体结构图,图 5为配位多面体图,图 6为化合物3的拓扑结构图。由图 4和图 5可知,该化合物为多齿配位化合物。在化合物3中,Mn有2种配位环境,Mn1与6个氧结合形成六配位的八面体结构,Mn2与5个氧原子结合形成六面体结构。在Mn1的配位环境中,O4、O5来自于水中的氧,O1、O3、O6H、O10来自BTCA4-羧基氧;在Mn2的配位环境中,O2、O7、O8、O9自于BTCA4-中的羧基氧,O11来自水中氧。由表 3可知,Mn-O的键长中最长为0.235 5 nm,最短为0.213 3 nm,与文献报道的含锰-羧基单元的化合物键长相符[24]。由图 6可以看出,3以Mn1和Mn2为金属节点,以BTCA4-为连接器,采用八连接方式,形成4, 8-c网络,其拓扑符号为{412; 612; 84}2{46}2。
 图 4
化合物3的晶体结构图(50%椭球概率)
Figure 4.
Crystal structure of compound 3 shown as 50% thermal ellipsoid probability
图 4
化合物3的晶体结构图(50%椭球概率)
Figure 4.
Crystal structure of compound 3 shown as 50% thermal ellipsoid probability
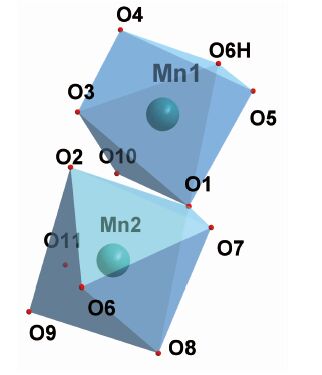 图 5
MnⅡ的配位多面体图
Figure 5.
Coordination polyhedron for MnⅡ
图 5
MnⅡ的配位多面体图
Figure 5.
Coordination polyhedron for MnⅡ
 图 6
化合物3的结构拓扑图
Figure 6.
Topological net of compound 3
图 6
化合物3的结构拓扑图
Figure 6.
Topological net of compound 3
2.2 红外光谱表征
配体H4BETA(a)与化合物1(b),2(c)的红外光谱如图 7所示。3种物质均在3 000 cm-1附近出现苯环C-H伸缩振动峰,在870~855 cm-1出现H4BETA四取代苯的C-H面外弯曲振动;H4BETA在3 560~ 3 500 cm-1出现羧基中的O-H伸缩振动吸收峰,化合物1中此峰位移至3 419.9 cm-1处,化合物2中此峰位移至3 550 cm-1处;H4BETA中在1 400 cm-1附近出现羧基中O-H弯曲振动吸收峰,在化合物1中此峰位移至1 397.5 cm-1,化合物2中此峰位移至1 383 cm-1;H4BETA在1 720 cm-1附近出现羧基中的C=O伸缩振动吸收峰,在化合物1中此峰位移至1 594.5 cm-1,化合物2中此峰位移至1 587 cm-1,表明羧基参与了钴离子的配位;化合物1与化合物2分别在659与820 cm-1附近出现Co-O键和Zn-O键吸收峰,进一步说明氧原子参与了配位[25-26]。
 图 7
化合物1、2与H4BETA的红外谱图
Figure 7.
Infrared spectra for compound 1, 2 and H4BETA
图 7
化合物1、2与H4BETA的红外谱图
Figure 7.
Infrared spectra for compound 1, 2 and H4BETA
配体H4BTCA(a)与化合物3(b)的红外光谱如图 8所示。由图可知,H4BTCA在3 300~2 500 cm-1出现羧基中的O-H伸缩振动吸收峰,在化合物3中此峰移至3 417 cm-1附近; H4BTCA在1 700~1 680 cm-1和950~890 cm-1出现C=O伸缩振动和羟基面外弯曲振动,化合物3中两峰分别移至1 573和 1 389 cm-1附近;化合物3在671 cm-1附近出现Mn-O键吸收峰,也说明氧原子参与了配位[27]。
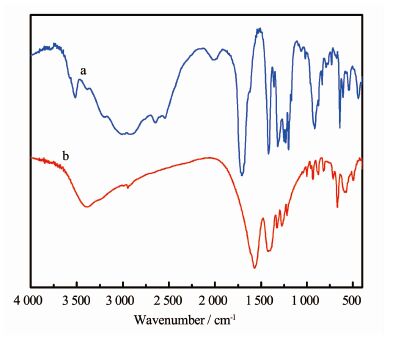 图 8
化合物3与H4BTCA的红外光谱图
Figure 8.
Infrared spectra for compound 3 and H4BTCA
图 8
化合物3与H4BTCA的红外光谱图
Figure 8.
Infrared spectra for compound 3 and H4BTCA
2.3 荧光性质测定
配体H4BETA(a,b)、化合物1(c,d)与化合物2(e,f)的荧光发射(Em)和激发(Ex)光谱如图 9所示。其中H4BTA的最大发射波长λEm=361 nm,这可以归属为π*→π的电子跃迁;化合物1和2的最大发射波长相同,λEm=315 nm,较配体发生了46 nm的蓝移,可能是因为配体与金属离子键合后,其扭转的构型限制了弛豫跃迁。化合物1和2的荧光光谱几乎重合,说明化合物的发光主要是基于配体本身的发光。配体H4BTA的最大激发波长λEx=319 nm,斯托克斯位移ΔV=42 nm;化合物1和2的最大激发波长λEx=284 nm,斯托克斯位移ΔV=31 nm,相较于配体,化合物1和2的斯托克斯位移更小,荧光效率更高。
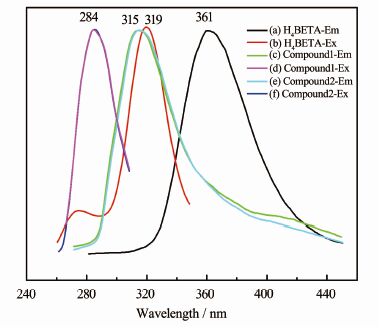 图 9
H4BETA (a, b)与化合物1(c, d),2 (e, f)的发射和激发光谱
Figure 9.
Emission and excitation spectra for H4BETA (a, b),compound 1(c, d ) and compound 2(e, f)
图 9
H4BETA (a, b)与化合物1(c, d),2 (e, f)的发射和激发光谱
Figure 9.
Emission and excitation spectra for H4BETA (a, b),compound 1(c, d ) and compound 2(e, f)
配体H4BTCA(a,b)与化合物3(c,d)的荧光发射(Em)和激发(Ex)光谱如图 10所示。其中H4BTCA的最大发射波长λEm=363 nm,这可以归属为π*→π的电子跃迁;化合物3的λEm=314 nm,较配体发生了49 nm的蓝移,可能是因为配体与金属离子键合后,其扭转的构型限制了弛豫跃迁;配体H4BTCA的最大激发波长λEx=272 nm,斯托克斯位移ΔV=91 nm,化合物3的最大激发波长λEx=285 nm,斯托克斯位移ΔV=29 nm,相较于配体,化合物3的斯托克斯位移更小,荧光效率更高。
 图 10
H4BTCA (a, b)与化合物3(c, d)的发射和激发光谱
Figure 10.
Emission and excitation spectra for H4BTCA (a, b) and compound 3 (c, d)
图 10
H4BTCA (a, b)与化合物3(c, d)的发射和激发光谱
Figure 10.
Emission and excitation spectra for H4BTCA (a, b) and compound 3 (c, d)
2.4 热稳性测定
化合物1的TG/DTG曲线如图 11(b)所示。由图可以看出,在100 ℃化合物1有失重,失重率是17.34%,这归属于化合物1中结晶水脱除(理论值 17.13%)。随后TG曲线下降缓慢,100至380 ℃之间失重7.1%,归属于配位水分子和羟基的失去(理论值 6.66 %)。当温度达到380 ℃后,化合物1的骨架开始分解,580 ℃左右时,化合物1失重完全,此时质量剩余率为26.93%,残渣主要为金属Co的氧化物(理论值28.04%),这部分失重可以归属于配体BETA4-的分解。与图 11(a)中的配体相比,H4BETA形成MOFs材料后,热稳定性有所提高。
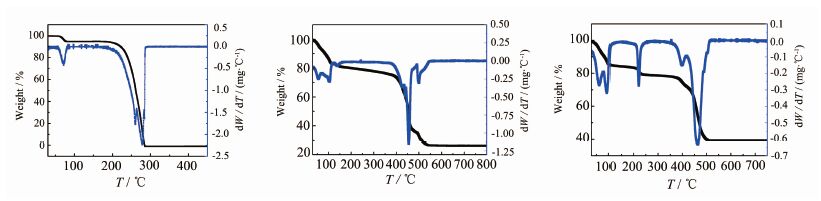 图 11
(a)H4BETA 的TG/DTG曲线;(b)化合物1的TG/DTG曲线;(c)化合物2的TG/DTG曲线
Figure 11.
(a) TG/DTG curves for H4BETA; (b) TG/DTG curves for compound 1; (c) TG/DTG curves for 2
图 11
(a)H4BETA 的TG/DTG曲线;(b)化合物1的TG/DTG曲线;(c)化合物2的TG/DTG曲线
Figure 11.
(a) TG/DTG curves for H4BETA; (b) TG/DTG curves for compound 1; (c) TG/DTG curves for 2
由于化合物2与1同构,其热稳定性也相似,其TG/DTG曲线如图 11(c)所示。由图可以看出从室温到100 ℃左右区间中,失重率为15.16%,这一部分的失重主要为化合物2中的结晶水脱除(理论值16.62%)。100~380 ℃,为化合物2的第2次失重,失重7.28%,归属为化合物2中的配位水分子和羟基的失去(理论值6.55%);当温度达到385 ℃左右时,化合物2的骨架开始瓦解,直至513 ℃左右时分解完全,此时剩余38.85%左右,残渣主要为ZnO(理论值37.56%)。H4BETA形成MOFs材料2后,热稳定性也有所提高。
化合物3的TG/DTG曲线如图 12(b)所示。由图可知,在室温至100 ℃之间失重率是4.96%,这可能是失去结晶水(理论值4.46%)。随后TG曲线出现缓慢下滑,在温度达到413 ℃左右时,迅速失重,当温度达到472 ℃左右时,质量下降的速率达到最大,此时质量分数为 64.83%左右,到800 ℃样品仍未分解完全,而H4BTCA在350 ℃左右基本分解完全(图 12(a))。
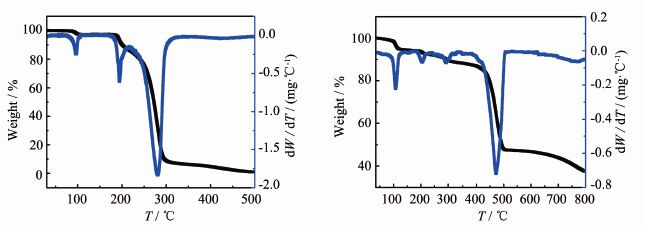 图 12
(a)H4BTCA 的TG/DTG曲线;(b)化合物3的TG/DTG曲线
Figure 12.
(a) TG/DTG curves for H4BTCA; (b) TG/DTG curves for compound 3
图 12
(a)H4BTCA 的TG/DTG曲线;(b)化合物3的TG/DTG曲线
Figure 12.
(a) TG/DTG curves for H4BTCA; (b) TG/DTG curves for compound 3
3 结论
本文选用H4BETA或H4BTCA为有机配体,合成得到3个结构新颖的MOFs材料。化合物1和2是同构的,是由五核金属簇连接有机配体BETA4-形成三维孔道结构,并具有一维孔道。化合物3由锰离子与BTCA4-连接形成框架结构,无明显的孔道。化合物1、2和3的荧光光谱研究表明其荧光主要是基于配体本身的发光,发射光谱的蓝移可能是因为配体与金属离子键合后,其扭转的构型限制了弛豫跃迁。同时,随着配体与金属离子间配位键的形成,增强了体系的刚性,提高了荧光的效率。
-
-
[1]
Li H L, Eddaoudi M, Yaghi O M, et al. Nature, 1999,402: 276-279 doi: 10.1038/46248
-
[2]
Janiak C, Vieth J K. New J. Chem., 2010,34(11):2366-2388 doi: 10.1039/c0nj00275e
-
[3]
Ferey G. Chem. Soc. Rev., 2008,37(1):191-214 doi: 10.1039/B618320B
-
[4]
Yuan D Q, Getman R B, Wei Z W, et al. Chem. Commun., 2012,48(27):3297-3299 doi: 10.1039/c2cc17168f
-
[5]
Wen L, Cheng P, Lin W B. Chem. Commun., 2012,48(23): 2846-2848 doi: 10.1039/c2cc17298d
-
[6]
Park H J, Suh M P. Chem. Commun., 2012,48(28):3400-3402 doi: 10.1039/c2cc17005a
-
[7]
Hou C, Liu Q, Lu Y, et al. Microporous Mesoporous Mater., 2012,152:96-103 doi: 10.1016/j.micromeso.2011.11.058
-
[8]
Yang J F, Yu Q H, Zhao Q, et al. Microporous Mesoporous Mater., 2012,161:154-169 doi: 10.1016/j.micromeso.2012.01.008
-
[9]
Eckhardt S, Brunetto P S, Gragnon J. Chem. Rev., 2013,113 (7):4708-4754 doi: 10.1021/cr300288v
-
[10]
Wang C, Volotskova O, Lu K D. J. Am. Chem. Soc., 2014, 136(17):6171-6174 doi: 10.1021/ja500671h
-
[11]
Deng H X, Doonan C J, Furukawa H, et al. Science, 2010,327 (5967):846-850 doi: 10.1126/science.1181761
-
[12]
Eddaoudi M, Kim J, Rosi N, et al. Science, 2002,295(5554): 469-472 doi: 10.1126/science.1067208
-
[13]
那立艳,姜慧明,杨宝灵.高等学校化学学报, 2007,28(8):1437-1439NA Li-Yan, JIANG Hui-Ming, YANG Bao-Ling, et al. Chem. J. Chinese Universities , 2007,28(8):1437-1439
-
[14]
Senkovska I, Hoffmann F, Froba M, et al. Microporous Mesoporous Mater., 2009,122(1/2/3):93-98
-
[15]
Liang Y C, Cao R, Su W P. Angew. Chem. Int. Ed., 2000, 39(18):3304-3307 doi: 10.1002/(ISSN)1521-3773
-
[16]
Jiang D M, Mallat T, Meier D M. J. Catal., 2010,270(1):26-33 doi: 10.1016/j.jcat.2009.12.002
-
[17]
Pau D G, Oscar F, Lauar C D, et al. Cryst. Growth Des., 2013,13(11):4735-4745 doi: 10.1021/cg4008679
-
[18]
Liu Y Y, Ma J F, Yang J, et al. CrystEngComm, 2008,10(7): 894-904 doi: 10.1039/b716476a
-
[19]
Sheldrick G M. SADABS, University of Göttingen, Germany, 1994.
-
[20]
Sheldrick G M. SHELX-97, Program for Crystal Structure Solution, University of Göttingen, Germany, 1997.
-
[21]
Sheldrick G M. SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997.
-
[22]
Zhang C L, Li Y L, Wang T, et al. Chem. Commun., 2015,51 (29):8338-8341
-
[23]
Cui S X, Zhang J P, Wang L, et al. Acta Crystallogr. Sect. E: Struct. Rep. Online, 2005,61(7):o2193-o2195 doi: 10.1107/S1600536805018593
-
[24]
张少华,杨颖群,匡云飞.无机化学学报, 2013,29(1):138-142 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20130121&flag=1ZHANG Shao-Hua, YANG Ying-Qun , KUANG Yun-Fei. Chinese J. Inorg. Chem., 2013,29(1):138-142 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20130121&flag=1
-
[25]
Zheng M X, Gao X J, Zhang C L, et al. Dalton Trans., 2015,44(10):4751-4758 doi: 10.1039/C4DT04011B
-
[26]
王锐.辽宁石油化工大学学报, 2007,27(4):24-27WANG Rui . J. Liaoning Shihua University , 2007,27(4):24-27
-
[27]
Kono S, Wang Y J, Tang L M. Polym. Adv. Technol., 2009,20(6):558-565 doi: 10.1002/pat.v20:6
-
[1]
-

图 1 化合物1的晶体结构图(50%椭球概率)
Figure 1 Crystal structure of the compound 1 shown as 50% thermal ellipsoid probability
Symmetry codes: A: 2-x, -y, 1-z; B: 1-x, -y, 1-z; C: x+1, y, z; E: 5 x-1, y, z; F: -x+5/2, y+1/2, -z+3/2; H: -x+5/2, y-1/2, -z+3/2

图 2 化合物1中CoⅡ的配位多面体图
Figure 2 Coordination polyhedron for CoⅡ in 1
Symmetry codes: A :2-x, -y, 1-z; B :1-x, -y, 1-z

图 3 化合物1和2的拓扑结构图
Figure 3 Topological net of compound 1 and 2
Symmetry codes: H: 0.5-x, -0.5+y, 0.5-z

图 4 化合物3的晶体结构图(50%椭球概率)
Figure 4 Crystal structure of compound 3 shown as 50% thermal ellipsoid probability
Symmetry codes: B : 0.5-x, 0.5+y, 0.5-z; D: -0.5-x, -0.5+y, 0.5-z; E :-0.5+x, 0.5-y, -0.5+z ; G :-x, 1-y, -z; H :0.5-x, -0.5+y, 0.5-z

图 7 化合物1、2与H4BETA的红外谱图
Figure 7 Infrared spectra for compound 1, 2 and H4BETA
a: H4BETA ; b: compound 1; c: compound 2

图 8 化合物3与H4BTCA的红外光谱图
Figure 8 Infrared spectra for compound 3 and H4BTCA
a: H4BTCA; b:compound 3

图 9 H4BETA (a, b)与化合物1(c, d),2 (e, f)的发射和激发光谱
Figure 9 Emission and excitation spectra for H4BETA (a, b),compound 1(c, d ) and compound 2(e, f)

图 10 H4BTCA (a, b)与化合物3(c, d)的发射和激发光谱
Figure 10 Emission and excitation spectra for H4BTCA (a, b) and compound 3 (c, d)

图 11 (a)H4BETA 的TG/DTG曲线;(b)化合物1的TG/DTG曲线;(c)化合物2的TG/DTG曲线
Figure 11 (a) TG/DTG curves for H4BETA; (b) TG/DTG curves for compound 1; (c) TG/DTG curves for 2

图 12 (a)H4BTCA 的TG/DTG曲线;(b)化合物3的TG/DTG曲线
Figure 12 (a) TG/DTG curves for H4BTCA; (b) TG/DTG curves for compound 3
表 2 化合物1的主要键长(nm)和键角(°)
Table 2. Selected bond lengths(nm) and bond angles(°) for compound 1
 下载: 导出CSV
下载: 导出CSV
表 3 化合物3的主要键长(nm)和键角(°)
Table 3. Selected bond lengths(nm) and bond angles (°) for compound 3
下载: 导出CSV
-













 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 2649
- HTML全文浏览量: 224
 下载:
下载:
