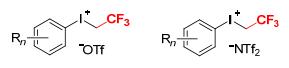
Figure 1.
General structures of aryl(trifluoroethyl)iodonium salts ([ArICH2CF3][X])

The incorporation of fluorine and fluorine-containing functionalities into organic compounds has become one of the most important strategies for the synthesis of new pharmaceuticals, agrochemicals, and functional materials.[1] Due to the unique stereoelectronic effects imparted by fluorine, bioactive organofluorides have exhibited advantageous properties such as good chemical stability, high lipophilicity, excellent membrane permeability, and good bioavailability compared to the nonfluorinated analogues.[1] The prevalence of fluorinated compounds in a wide range of research areas has motivated great interest in the development of efficient and convenient synthetic methods.[2] Among all types of organofluorides, the trifluoroethyl-containing organic small molecules have drawn increasing attention because of the special physicochemical and biological characteristics given by the trifluoroethyl groups (CH2CF3).[3] The direct trifluoroethylation methods with the readily available CH2CF3 reagents such as CF3CH2X (X=I, Br, OTs), CF3CHCl2, CF3CH2SO2Y (Y=Cl, Na), [ArICH2CF3][OTf], CF3CO2H, and CF3- CH2NH2 (or CF3CHN2) have been well documented in the last few decades.[3]
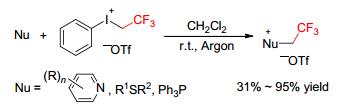
In particular, trifluoroethylation using aryl(trifluoroethyl)iodonium salts ([ArICH2CF3][X], Figure 1) as CH2CF3 transfer sources has attracted great interest. These reagents have showed much powerful electrophilic trifluoroethylation reactivity toward a variety of nucleophiles.[4~7] They can readily construct the crucial N-CH2CF3, O-CH2CF3, C-CH2CF3, S-CH2CF3, and P-CH2CF3 bonds for numerous biologically active molecules in diverse reactions, showing much higher electrophilicity than the other CH2CF3 reagents. In this review, we summarize the trifluoroethylation reactions using aryl(trifluoroethyl)-iodonium salts as the CH2CF3 sources, which are divided into three parts. The first part describes the synthesis of aryl(trifluoroethyl)iodonium salts. The second and third parts discuss the transition metal-free and metal-catalyzed trifluoroethylation reactions, respectively, using [ArICH2-CF3][X] reagents. This review will work as a reference for both younger and established researchers to gain an insight into the areas of trifluoroethylation using aryl(trifluoroethyll)iodonium salts.
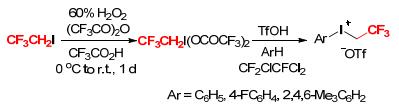
Aryl(trifluoroethyl)iodonium triflates were first synthesized by Umemoto and Gotoh in the 1980s (Scheme 1).[4] In the first step of their synthesis, reaction of 1, 1, 1-trifluoro-2-iodoethane (CF3CH2I) at 0 ℃ to room temperature with trifluoroperacetic acid, which was prepared in situ by adding 60% hydrogen peroxide to trifluoroacetic anhydride in the presence of a catalytic amount of trifluoroacetic acid, gave quantitatively the key intermediate (2, 2, 2-trifluoroethyl)-iodanediyl bis(2, 2, 2-trifluoroacetate). In the second step, treatment of (2, 2, 2-trifluoroethyl)- iodanediyl bis(2, 2, 2-trifluoroacetate) with a series of arenes (ArH) in 1, 1, 2-trichloro-trifluoroethane (CF2ClCF- Cl2) in the presence of triflic acid (TfOH) afforded aryl(trifluoroethyl)iodonium triflates ([ArICH2CF3][OTf]) in good yields.[4]
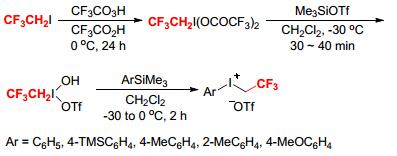
Later, Zhdankin and coworkers disclosed another useful approach for the production of aryl(trifluoroethyl)iodomum triflates. In their procedures, 1-(hydroxy(sulfonyloxy)iodo)-2, 2, 2-trifluoroethanes [CF3CH2I(OH)OSO2R (R=CH3, CF3, p-CH3C6H4)] were synthesized as the intermediates by oxidation of CF3CH2I with trifluoroperacetic acid followed by reaction with TsOH, MsOH, or Me3Si- OTf (Scheme 2).[5] Then, reactions of CF3CH2I(OH)OTf with (trimethylsilyl)arenes provided the desired aryl(trifluoroethyl)iodomum triflates ([ArICH2CF3][OTf]).
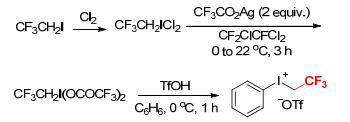
In the above methods, the first step for the synthesis of [ArICH2CF3][OTf] was the oxidation of CF3CH2I to the bis(trifluoroacetoxy)iodonium compound by 60%~80% H2O2 in (CF3CO)2O.[6] However, hydrogen peroxide of this grade was not commonly available, and the optimal procedure required a large amount of the expensive anhydride. To solve these problems, Montanari and coworker reported a two-step protocol for conversion of 1-(dichloroiodo)- 2, 2, 2-trifluoroethane (CF3CH2ICl2) to [PhICH2CF3][OTf] (Scheme 3).[6] CF3CH2ICl2, which was simply derived from CF3CH2I and chlorine, reacted with silver trifluoroacetate in CFCl2CF2Cl (CFC-113) at 0 to 22 ℃, followed by filtration on a glass flit, to provide a clear or pale yellow solution of CF3CH2I(OCOCF3)2. This solution was treated with benzene in the presence of TfOH at 0 ℃ to afford [PhICH2CF3][OTf] as a white nonhygroscopic powder in good yield. In this procedure, chlorine and nontoxic CFC-113 replaced 60%~80% H2O2 and trifluoroacetic anhydride as the oxidant and the solvent, respectively, in the synthesis of the key CF3CH2I(OCOCF3)2 intermediate.[6]
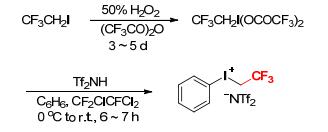
Phenyl(trifluoroethyl)iodonium triflate ([PhICH2CF3]- [OTf]) can not be used in aqueous media or basic solvents as it is instantly destroyed under these conditions.[7] DesMarteau and coworker synthesized phenyl(trifluoroethyl)iodonium bis(trifluoromethanesulfonyl)amide ([PhI-CH2CF3][NTf2]) from CF3CH2I and (CF3SO2)2NH, which is slightly soluble in water and is hardly hydrolyzed (Scheme 4).[7] The key intermediate CF3CH2I(OCOCF3)2 was similarly constructed by oxidation of CF3CH2I with 50% H2O2 in (CF3CO)2O at room temperature for 3~5 d. Subsequently, addition of Tf2NH to a solution of CF3CH2I(OCOCF3)2 in CFC-113 followed by treatment with benzene at 0 to 25 ℃ gave [PhICH2CF3][NTf2] as a white powder in good yield. The preparation of [PhICH2CF3][NTf2] is simple, but the anhydrous conditions must be maintained throughout the whole procedure.[7]
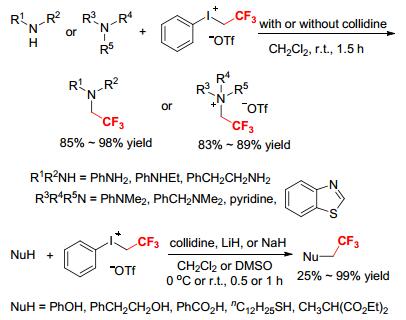
Aryl(trifluoroethyl)iodonium salts have been widely used as electrophilic trifluoroethylation reagents in organic synthesis since their first preparation. In 1986, Umemoto and Gotoh disclosed an efficient and practicable protocol for the transition metal-free trifluoroethylation of different types of nucleophiles by [PhICH2CF3][OTf] (Scheme 5).[8] The reactions proceeded at room temperature under mild conditions to give the corresponding trifluoroethylated products in varying yields. For instance, reactions of [PhICH2CF3][OTf] with aniline, N-ethylaniline, and phenylethylamine provided N-trifluoroethyl amines in high yields.[8] Treatment of [PhICH2CF3][OTf] with lithium phenoxide and lithium phenylethoxide at 0 ℃ afforded 79% of (2, 2, 2-trifluoroethoxy)benzene and 61% of (2- (2, 2, 2-trifluoroethoxy)ethyl)benzene, respectively (Scheme 5).[8] When [PhICH2CF3][OTf] reacted with benzoic acid in the presence of collidine (1 equiv.), 2, 2, 2-trifluoroethyl benzoate was formed in 99% yield. [PhICH2CF3][OTf] treated with mercaptan without base produced the expected trifluoroethyl thioether in good yield. The reactions of [PhICH2CF3][OTf] with diethyl methylmalonate and trimethyl((1-phenylvinyl)oxy)silane provided the respective trifluoroethylated products in low yields. Treatment of [PhICH2CF3][OTf] with tertiary amines or N-heterocyclic compounds supplied N-trifluoroethyl ammonium or pyridinium triflates in good yields (Scheme 5).[8] Furthermore, aniline reacted with 3 equivalents of [PhICH2CF3][OTf] in the presence of 2, 6-di-t-butyl-4-methylpyridine at room temperature for 2 h to furnish N, N-bis(2, 2, 2-trifluoroethyl)- aniline in 94% yield, suggesting that bis(trifluoroethylation) of primary amines by [PhICH2CF3][OTf] was feasible in a one-pot procedure.[8]
The same strategy was then applied to the trifluoroethylation of pyridine derivatives, quinoline, benzothiazole, sulfides, dibenzothiophene, and triphenylphosphine by Umemoto and Gotoh again, affording a series of the corresponding trifluoroethylated onium salts in good yields at room temperature (Scheme 6).[9] Diphenyl sulfoxide and dimethyl sulfoxide were also smoothly converted by [PhICH2CF3][OTf], but the desired O- or S-trifluoroethyl salts were not isolated in a pure form because of their hygroscopic nature. Pyridine N-oxide reacted mildly with [PhICH2CF3][OTf] to form N-(2, 2, 2-trifluoroethoxy)pyridinium triflate in a quantitative yield.
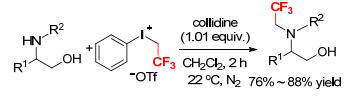
In a similar manner, Montanari and coworker studied the N-trifluoroethylation of aminoalcohols by phenyl(trifleoroethyl)iodonium triflate using 2, 4, 6-collidine as a base (Scheme 7).[6] The trifluoroethylation occurred selectively at the nitrogen centers rather than the oxygen sites, which proceeded fast and did not require protection of the hydroxy groups.
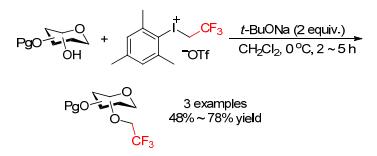
In 2016, Olofsson and coworkers developed an interesting method for O-functionalization of carbohydrate derivatives using the bench-stable and easily prepared iodonium(Ⅲ) reagents (Scheme 8).[10] Mesityl(trifluoroethyl)- iodonium triflate ([MesICH2CF3][OTf]) was found effective to trifluoroethylate a small series of mono-unprotected sugar derivatives (e.g., furanose and pyranose). The reactions took place at 0 ℃ with complete chemoselectivity to give the 2, 2, 2-trifluoroethylated carbohydrates under mild conditions. This protocol was the first time introducing trifluoroethyl groups onto carbohydrate hydroxyl moieties, thus attaining products that were previously inaccessible or required long synthetic sequences due to the low electrophilicity of the other CH2CF3 reagents.
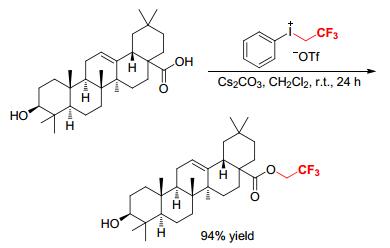
Recently, Zhang and coworkers described a facile and efficient method for direct trifluoroethylation of long-chain aliphatic acids by [PhICH2CF3][OTf] in the presence of Cs2CO3 (Scheme 9).[11] A large number of complex substrates were smoothly converted at room temperature without other additives to afford the corresponding 2, 2, 2-trifluoroethyl aliphatic acid esters in excellent yields. Compared to the known traditional synthetic methods for trifluoroethyl esters, the reaction did not use harsh and toxic condensation agents, and thus, the carbon-carbon double bonds, the free hydroxyl group, and the amido functionalities in substrates were all tolerated. This protocol offered a green access to 2, 2, 2-trifluoroethyl aliphatic acid esters.
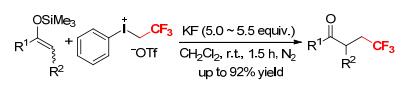
Aryl(2, 2, 2-trifuoroethyl)iodonium salts are also useful reagents in the formation of C—CH2CF3 bonds.[12] Umemoto and Gotoh found that the enol trimethylsilyl ethers of carbonyl compounds reacted with [PhICH2CF3]- [OTf] in CH2Cl2 in the presence of a large access of potassium fluoride (KF) at room temperature to give α-trifluoroethylated carbonyls in good yields (Scheme 10).[12] Likewise, an enol trimethylsilyl ether of α, β-unsaturated ketone reacted with [PhICH2CF3][OTf] to form a γ-trifluoroethylated product accompanied by a trace amount of α-trifluoroethylated compound, showing good selectivity of the reaction. KF successfully promoted the reactions of enol trimethylsilyl ethers with [PhICH2CF3]- [OTf] by attacking the silyl sites of the enol ether moieties of the enol silyl ethers.
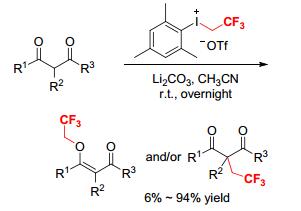
A transition metal-free, substrate-dependent selective O- or C-trifluoroethylation of 1, 3-dicarbonyls by [MesICH2CF3][OTf] was reported under mild conditions (Scheme 11).[13] Various 1, 3-dicarbonyls reacted with [MesICH2CF3][OTf] at room temperature in the presence of an appropriate base to give O-trifluoroethylated products, C-trifluoroethylated products, or a mixture of O- and C-trifluoroethylated products in good yields. The product type was dramatically dependent upon the structure of the starting 1, 3-dicarbonyls in this reaction. The cyclic 1, 3-diketones exclusively afforded the O-trifluoroethylated products, whereas the acyclic 1, 3-diketones, β-keto esters, and malonates selectively or specifically formed the C-trifluoroethylated products. Li2CO3 facilitated the C-trifluoroethylation of acyclic 1, 3-diketones and β-keto esters. The reaction did not require pre-activation of 1, 3-dicarbonyls and use of strong bases.
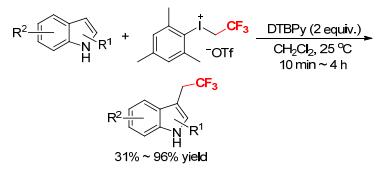
A transition metal-free direct C—H trifluoroethylation of indoles by [MesICH2CF3][OTf] was explored by Novák and coworkers (Scheme 12).[14] The reaction occurred at the C3 position of indole cores within 10~240 min to afford the corresponding trifluoroethylated products in good yields. The density functional theory (DFT) calculations of the reaction revealed that dissociation of the triflate anion from [MesICH2CF3][OTf] is slightly exergonic (-0.9 kcal•mol-1), suggesting a dissociated resting state, and that the rate-determining step of the reaction is the trifluoroethylation of the indole ring.[14] To explain the special efficiency of DTBPy as a base in this reaction, the activation barrier of the N-trifluoroethylation of DTBPy by [MesICH2CF3][OTf] was calculated in comparison with the other amines such as TEA, DIPEA, and 2, 6-lutidine.[14] The results showed that N-trifluoroethylation of DTBPy required extremely high activation energy due to the significant steric repulsion, and that the activation barrier differences between the competing N- and C-trifluoroethylations determined the efficiency of the substrate/base combinations. This reaction offered a new access to the trifluoroethyl heteroaromatic compounds via C—H functionalization.
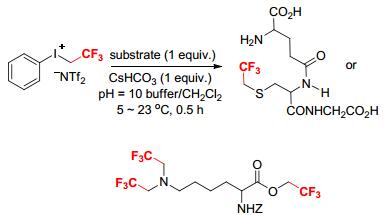
In contrast to [PhICH2CF3][OTf], [PhICH2CF3][NTf2] is stable to water.[7] DesMarteau and coworker described the first aqueous trifluoroethylation reaction using [PhICH2CF3][NTf2] as the trifluoroethylation reagent. They found that reactions of amino acids and peptides with [PhICH2CF3][NTf2] in the presence of CsHCO3 in a degassed mixture of buffer (Hydrion, Na2CO3-NaHCO3, pH=10) and CH2Cl2 at 5~23 ℃ for 0.5 h afforded the corresponding trifluoroethylated products in good yields (Scheme 13).[7]
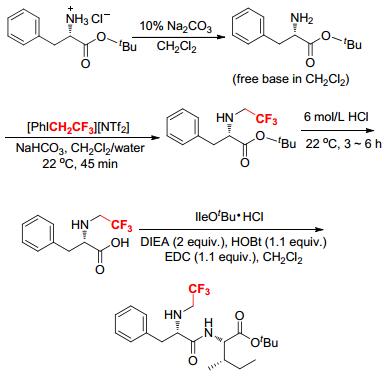
Similarly, Nα-trifluoroethyl-phenylalanine, Nα-trifluoroethyll-tyrosine and Nα-trifluoroethyl-valine were synthesized from the reactions of [PhICH2CF3][NTf2] with the corresponding amino acids at the α-nitrogen in a two-phase solvent system (CH2Cl2/water/NaHCO3) (Scheme 14).[15] The standard coupling of these Nα-trifluoroethyl amino acids gave twelve novel unnatural peptides in high yields without racemization. Nα-Trifluoroethyl amino acids could behave as N-protected amino acids.[15] The α-nitrogen of Nα-trifluoroethyl amino acids was essentially non-nucleophilic because of the presence of the sterically hindered and electron-withdrawing CH2CF3 groups. This unexpected property promised a large number of potentially bioactive fluoroalkylated substances from Nα-trifluoroethyl amino acids that possess stable and lipophilic moieties.[15]
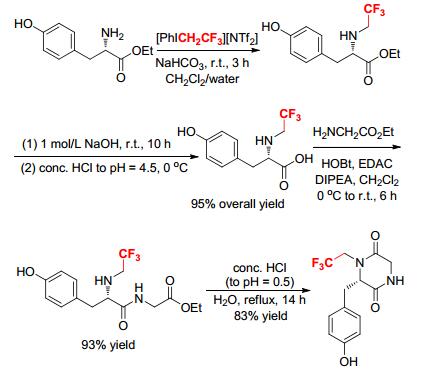
The unexpected formation of 3-(S)-[(4-hydroxyphenyl)methyl]-4-[(2, 2, 2-trifluoro)ethyl]-2, 5-piperazinedione and its analogues via intramolecular cyclization at the nitrogen atoms of Nα-trifluoroethylated amino acids was reported (Scheme 15).[16] Initially, reaction of L-tyrosine ethyl ester with [PhICH2CF3][NTf2] in a solvent mixture of CH2Cl2/H2O produced Nα-trifluoroethylated L-tyrosine ethyl ester, which was hydrolyzed under basic conditions followed by acidification with HCl to give Nα-trifluoroethylated L-tyrosine. Then, coupling of Nα-trifluoroethylated L-tyrosine with glycine ethyl ester in a solution phase yielded a linear dipeptide ester (Nα-CF3CH2-L-TyrGly-OEt). Cyclization of Nα-CF3CH2- L-Tyr-Gly-OEt in acidic aqueous solution at reflux gave piperazine-2, 5-dione in 83% yield. Analogously, several other trifluoroethylated cyclic dipeptides were also prepared in good yields (82%~95%) from the corresponding linear Nα-trifluoroethylated dipeptides L-TyrXOR (X=D-Ala, L-Leu, L-Phe, L-Glu; R=H, Me, Et).[16] These results demonstrated that the trifluoroethylated N-termini in linear dipeptides could undergo amide bond formation through intramolecular cyclization in acidic aqueous solutions.[16]
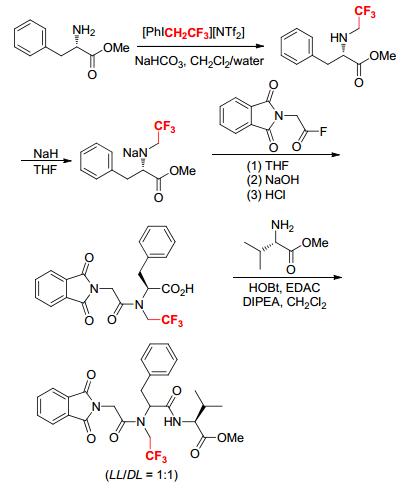
The α-amino group of L-phenylalanine methyl ester was trifluoroethylated by [PhICH2CF3][NTf2] in a solvent mixture of CH2Cl2/H2O, leading to Nα-trifluoroethyl L-phenylalanine methyl ester (Scheme 16).[17] Deprotonation of Nα-trifluoroethyl L-phenylalanine methyl ester at the α-amino position followed by coupling with Nα-phthaloyl glycine acid fluoride formed a trifluoroethylated dipeptide (Gly-(L)Phe), which further reacted with L-leucine methyl ester under the conventional carboxyl activation conditions to provide two diastereomers of the tripeptide Gly- (D, L)Phe-(L)Leu. Full racemization at the α-carbon site might be attributed to the electron-withdrawing CH2CF3 group at the adjacent nitrogen. The presence of CH2CF3 group might also facilitate the peptide bond rotation, resulting in different conformations, for that the CH2CF3 group decreased the double bond character of the amide and made the rotation easier.[17]
In 2003, DesMarteau and coworkers reported the direct trifluoroethylation of pyridine derivatives with [PhI-CH2CF3][NTf2] (Scheme 17).[18a] The reactions proceeded smoothly under mild conditions to furnish several room temperature ionic liquids in good yields. Five years later, the same research group disclosed a facile N-trifluoroethylation of L-histidine methyl ester with a large excess of [PhICH2CF3][NTf2] in CH2Cl2/H2O, which interestingly formed a triply trifluoroethylated ionic salt in 91% yield (Scheme 17).[18b] The extension of this reaction for 1-methyl-1H-imidazole resulted in an imidazolium-based ionic liquid. The reaction for N-1H, 1H-perfluoroalkylation of N-methyl imidazole was likely via an SN2 process. The lone pair electrons on the nitrogen atom interact with the empty antibonding orbital between the iodine and the sp3 carbon (CH2) to weaken and break the corresponding bonding orbital. With leaving of iodobenzene, the new σ-bond between the nitrogen atom and the sp3 carbon is formed to give the final product. Compared with the iodides RfnCH2I, this process showed the increased SN2 reactivity with the hypervalent iodonium salts.[18]
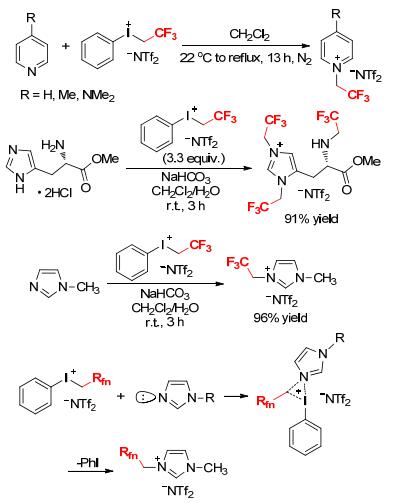
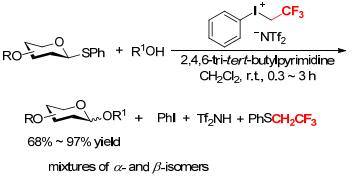
The use of phenyl(trifluoroethyl)iodonium bis(trifluoromethanesulfonyl)amide ([PhICH2CF3][NTf2]) as a water- and air-stable promoter for facile thioglycoside activation was described by Bennett and coworkers (Scheme 18).[19] Both armed and disarmed thioglycosides underwent rapid glycosylation with acceptors R1OH to form the desired products in 68%~97% yield. The reaction proceeded at room temperature in dichloromethane (DCM) in the presence of 2, 4, 6-tri-tert-butylpyrimidine (TTBP) under mild conditions and did not require strict exclusion of air and moisture. [PhICH2CF3][NTf2] was stable for 5 d at room temperature and could be kept for more than 6 months if stored in a refrigerator in the darkness.[19] This reagent offered an advantage over other glycosylation promoters through its particular ease of handling.
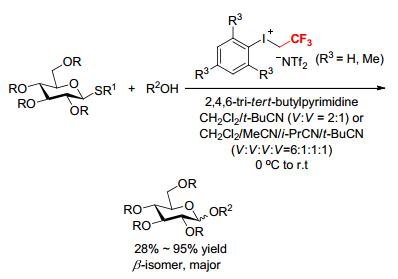
Short after, the direct glycosylation of armed thioglycoside derivatives by acceptors (R2OH) using [ArICH2CF3]- [NTf2] as a promoter was explored by Bennett's research group again (Scheme 19).[20] In this study, various combinations of solvents were examined, and both the solvent and the iodonium promoter were found to impact the stereoselectivity of the reaction. A combination of aryl(trifluoroethyl)iodonium bis(trifluoromethanesulfonyl)- amide and either a mixture of CH2Cl2 and pivalonitrile (V:V=2:1) or a mixture of CH2Cl2, acetonitrile, isobutyronitrile, and pivalonitrile (V:V:V:V=6:1:1:1) permit the glycosylation with moderate to excellent selectivity (up to 25:1 β/α). The reaction was conducted at 0 ℃ to room temperature, a much higher temperature than normally required to take advantage of the nitrile effect.[20] Given the operational simplicity of the process and the stability of all reagents involved, this reaction is useful for researchers to produce their target oligosaccharides with minimal synthetic training.
The introduction of trifluoroethyl groups into organic frameworks is an important reaction in organic chemistry.[3, 21] Although the straightforward trifluoroethylation at the carbon centers to form C—CH2CF3 bonds can be achieved by transition metal-catalyzed cross-coupling reactions, such transformation still represents a highly challenging synthetic task. So far, the copper-, cobalt-, nickel-, and palladium-catalyzed reactions using trifluoroethyl halides, tosylates, and diazo compounds have enabled trifluoroethylation of various aromatic systems.[3a, 21] The direct installation of trifluoroethyl groups into aromatic rings by transition metal-catalyzed C—H activation have also been realized.[21] Despite that these state-of-the-art trifluoroethylation methods have demonstrated the possibility of the direct incorporation of trifluoroethyl groups into unactivated arenes, the narrow substrate scopes, the harsh reaction conditions, the complicated reaction mixtures, and the occasional selectivity problems have demanded for further methodology development. The recent studies have showed that utilization of a highly active trifluoroethylation reagent can somewhat solve these problems, offering new synthetic opportunities for direct trifluoroethylation under transition-metal catalysis.
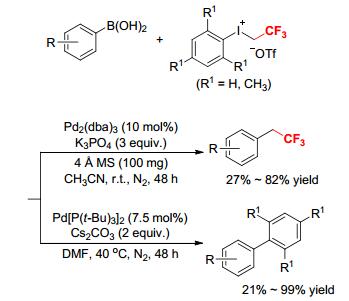
In 2016, Zhang and coworkers disclosed a Pd-catalyzed divergent trifluoroethylation and arylation of arylboronic acids by aryl(trifluoroethyl)iodonium triflates in the presence of different catalysts, bases and solvents (Scheme 20).[22] The nature of Pd-catalysts, bases, and solvents dramatically influenced the outcomes of the reaction. When the reactions of electron-rich arylboronic acids with [PhICH2CF3][OTf] or [MesICH2CF3][OTf] were performed in CH3CN in the presence of Pd2(dba)3 and K3PO4 at room temperature, the trifluoroethylation products were obtained in up to 82% yields. While if the reactions of electron-rich and electron-poor arylboronic acids with aryl(trifluoroethyl)iodonium triflate were run in DMF in the presence of Pd[P(t-Bu)3]2 and Cs2CO3 at 40 ℃, the arylation products were obtained in up to 99% yields. This tunable protocol represented the first use of aryl(2, 2, 2-trifluoroethyl)iodoniums as both trifluoroethylation and arylation reagents in the Pd-catalyzed reactions, allowing for an efficient and convenient access to trifluoroethyl arenes and biaryls under mild conditions without additional ligands.[22]
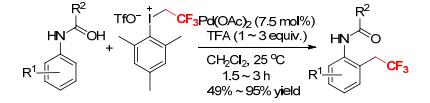
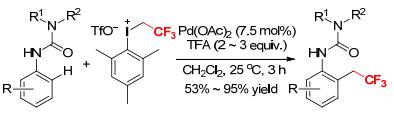
A simple Pd-catalyzed C—H trifluoroethylation of anilides using mesityl(trifluoroethyl)iodonium triflate as CH2CF3 reagent was described by Novák and coworkers (Scheme 21).[23] The preliminary study showed that the stoichiometric reaction of mesityl(trifluoroethyl)iodonium triflate with the trifluoroacetate-bridged dimeric complex of 2-methylacetanilide and palladium rapidly formed the ortho-trifluoroethylated product in 76% yield.[23] Based on this, the catalytic procedures were investigated. Various anilides reacted with [MesICH2CF3][OTf] at 25 ℃ for 1.5~3 h in the presence of 1~3 equivalents of 2, 2, 2-trifluoroacetic acid (TFA) to afford the desired ortho-trifluoroethylated products in good yields.[23] The reaction exhibited high selectivity and tolerated a broad range of functional groups.
In a similar manner, Novák and coworkers developed a palladium-catalyzed trifluoroethylation process for the preparation of ortho-trifluoroethyl aromatic ureas through C—H activation (Scheme 22).[24] The use of [MesICH2CF3][OTf] as a CH2CF3 transfer source allowed the efficient and selective introduction of the trifluoroethyl group at 25 ℃. 2, 2, 2-Trifluoroacetic acid (TFA) significantly improved the formation of the desired products. The DFT calculations suggested that the rate determining oxidative CH2CF3 transfer step was preceded by an unexpected C—H activation route on the Pd center during the catalytic cycle, where the deprotonation was assisted by an external triflate anion.[24] The formation of bimetallic palladium species is a crucial step of the reaction, which has an important role in the mild catalytic transformation. This reaction accomplished direct and late-stage trifluoroethylation of urea derivatives with good yields, high efficiency, and excellent functional group tolerance under mild conditions.[24]
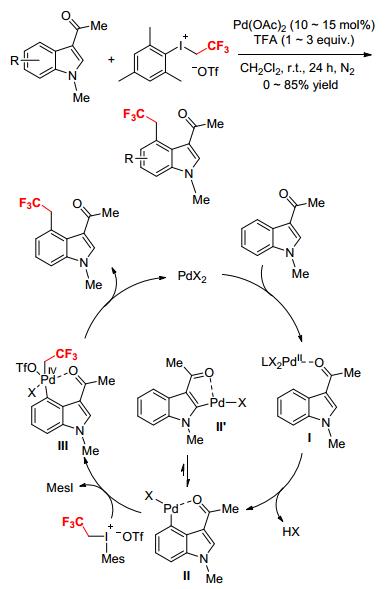
A mild synthetic protocol for direct installation of CH2CF3 groups at the C4-position of indoles by Pd-catalyzed C—H activation using [MesICH2CF3][OTf] as a highly reactive trifluoroethylation reagent was explored (Scheme 23).[25] The key to the high regioselectivity of the reaction is the appropriate choice of an easily accessible and removable directing group at the C3 position of indoles in the presence of Pd(OAc)2. The reaction provided moderate to high yields of the trifluoroethylated products and exhibited good functional group tolerance. Indoles bearing electron-donating groups at the C2, C6, or C7 positions underwent facile trifluoroethylation to afford the desired products in good yields. This strategy was also suitably utilized in the C4-trifluoroethylation of 3-benzo[b]thiophene, affording the desired product in 80% yield. Mechanistically, a plausible catalytic cycle was suggested for the reaction (Scheme 23).[25] First, palladium species coordinates with the acetyl group of the substrate to form complex Ⅰ, which activates the C—H bond at the C4 position of indole to generate intermediate Ⅱ with the release of HX. Then, oxidative addition of Ⅱ with [MesICH2CF3][OTf] affords a PdⅣ intermediate (Ⅲ), which finally undergoes reductive elimination to yield C4-trifluoroethylation products and regenerates the active PdⅡ species. The acetyl directing group at the C3 position was crucial to the high regioselectivity of the reaction, as it significantly affected the relative stabilities of the key intermediates Ⅱ and Ⅱ' in the catalytic process.
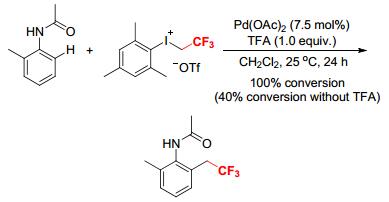
A Pd-catalyzed direct trifluoroethylation of aromatic amides with [MesICH2CF3][OTf] at room temperature under acidic conditions was developed by Loh and coworkers (Scheme 24).[26] The reaction was applicable to a variety of aromatic amides, and tolerated many functional groups, giving the corresponding ortho-trifluoro-ethylated products in good to high yields. Further studies showed that the trifluoroethylation was greatly affected by the steric hindrance of the substituents on the aryl rings of amides. The N—H bond on the amide nitrogen atom was indispensable for the transformation. A dimeric palladacycle, synthesized from the reaction of N-adamantyl-substituted benzamide with Pd(OAc)2/TFA, reacted with [MesCH2CF3][OTf] in the absence of TFA to afford the desired ortho-trifluoroethylated product in 80% yield, supporting a direct C—H alkylation pathway and a cyclopalladated intermediate and specifying the role of TFA in the reaction (Scheme 24).[26] Moreover, the intramolecular isotopic studies with an equimolar mixture of N-methylbenzamide and [D5]-N-methylbenzamide gave a kinetic isotopic effect (KIE) of 1.98, implying that the breakage of the C—H bond is a kinetically relevant step in the mechanistic profile. The recovered starting material N-methylbenzamide did not show any significant H/D exchange, which is an indicator of a largely irreversible cyclometalation. Based on these, a plausible reaction mechanism was suggested (Scheme 24).[26] The protic acid TFA first activates the palladium catalyst through ligand exchange, and the activated species (Pd(TFA)n) undergoes C—H insertion assisted by the directing group to form a cyclometalated intermediate (Ⅳ) or a N-palladated species (Ⅳ'). The strong oxidizing ability of the hypervalent iodonium reagent [MesCH2CF3][OTf] enables the conversion of intermediate Ⅳ to a PdⅣ complex (Ⅴ). Finally, reductive elimination of Ⅴ furnishes the product and regenerates the active PdⅡ species for the next catalytic cycle.
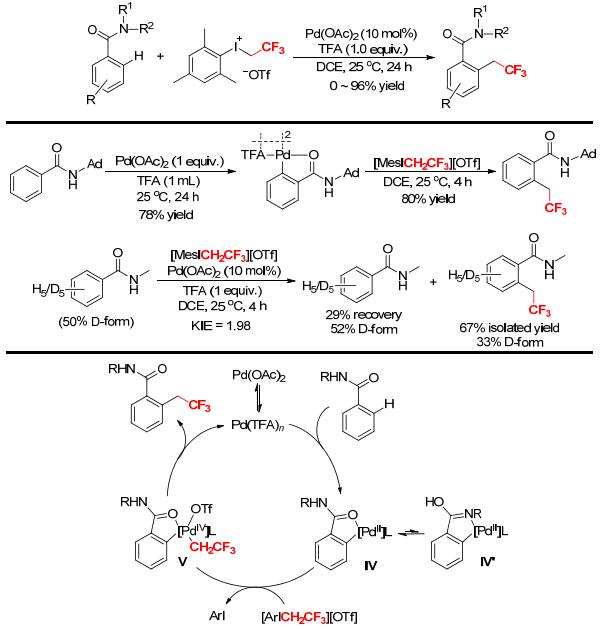
It was known that the additive CF3CO2H (TFA) could significantly promote the transition metal-catalyzed trifluoroethylation reactions and greatly increase the yields of the products. However, the roles of CF3CO2H in these reactions remained unclear. Recently, the Pd(OAc)2-catalyzed trifluoroethylation of N-(o-tolyl)acetamide with [MesICH2CF3][OTf] was chosen as a model reaction to explore the reaction mechanism and the roles of CF3CO2H by using the density function theory (DFT) computations (Scheme 25).[27] The results revealed that the whole catalytic cycle involved a coordination process of Pd(OAc)2 with N-(o-tolyl)acetamide, C—H bond activation, and a two-step migration of the CH2CF3 group. In the C—H activation step, the CH3COO‒ anion has stronger proton abstraction ability than the other species. The two-step migration of CH2CF3 group from [MesICH2CF3][OTf] to the product is more favorable than the direct transfer pathway. Moreover, CF3CO2H reacts with [MesICH2CF3][OTf] to form a new active species mesityl-(trifluoroethyl)iodonium trifluoroacetate ([MesICH2CF3][OCOCF3]), which facilitates the cleavage of the C—H bond, decreases the rate-limiting free energy barrier of the reaction, and changes the rate-determining step from the transfer of CH2CF3 group to the activation of C—H bond.[27]
In summary, the recent years have witnessed the significant progress on the transition metal-catalyzed or metalfree trifluoroethylation reactions using aryl(trifluoroethyl)- iodonium salts as the trifluoroethylation reagents. Numerous amines, peptides, phenols, thiols, sulfides, phosphines, carbonyl compounds, saccharides, glucosides, aliphatic acids, and electron-rich arenes were smoothly converted in these reactions to form the corresponding trifluoroethylated products in good yields. The use of the highly reactive aryl(trifluoroethyl)iodonium salts and the survey of the effective reaction systems are the key factors for the above achievements. Advantages of these reactions include simplicity, mild reaction conditions, high efficiency, good functional group tolerance, and a wide range of substrates. In addition, it is noteworthy that aryl(trifluoroethyl)iodonium bis(trifluoromethanesulfonyl)amides have been confirmed as the robust CH2CF3 sources for the functionalization of amino acids and peptides in aqueous media.
Although breakthroughs have been made in the transition metal-free or metal-catalyzed trifluoroethylation reactions, significant improvements are still necessary for these conversions. Trifluoroethylation of the least nucleophilic compounds under transition metal-free conditions is always a big challenge in organic synthesis. Extension of the transition metal-catalyzed trifluoroethylation methods for diverse carbon nucleophiles beyond aromatic systems is regarded in high demand. Since the commonly used reagents such as CF3CH2X (X=I, Br, OTs, OTf), CF3CHCl2, and CF3CH2SO2Y (Y=Cl, Na) have usually showed poor reactivities in the trifluoroethylation of target molecules, the utilization of the powerful aryl(trifluoroethyl)iodonium salts would be increasingly enhanced. Besides, another question arising is whether the direct asymmetric trifluoroethylation can be performed by using aryl(trifluoroethyl)- iodonium salts as the CH2CF3 reagents. We hope that this review will provide a reference and an appeal for the future researchers to develop new trifluoroethylation reactions for broader ranges of substrates, especially the late-stage trifluoroethylation of complicated molecules, with the promising aryl(trifluoroethyl)iodonium salts.
(a) Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed., Wiley-VCH, Weinheim, 2013.
(b) Gagnon, M.-C.; Auger, M.; Paquin, J.-F. Org. Biomol. Chem. 2018, 16, 4925.
(c) Nosova, E. V.; Lipunova, G. N.; Charushin, V. N.; Chupakhin, O. N. J. Fluorine Chem. 2018, 212, 51.
(d) Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887.
(e) Meanwell, N. A. J. Med. Chem. 2018, 61, 5822.
(f) Wang, B.-C.; Wang, L.-J.; Jiang, B.; Wang, S.-Y.; Wu, N.; Li, X.-Q.; Shi, D.-Y. Mini-Rev. Med. Chem. 2017, 17, 683.
(a) Uneyama, K.; Yamazaki, T. J. Fluorine Chem. 2017, 203, 3.
(b) Yang, L.; Dong, T.; Revankar, H.-M.; Zhang, C.-P. Green Chem. 2017, 19, 3951.
(c) Zhang, P.; Lv, L.; Shen, Q. Acta Chim. Sinica 2017, 75, 744 (in Chinese).
(张盼盼, 吕龙, 沈其龙, 化学学报, 2017, 75, 744.)
(d) Eisenstein, O.; Milani, J.; Perutz, R. N. Chem. Rev. 2017, 117, 8710.
(e) Song, H. X.; Han, Q. Y.; Zhao, C. L.; Zhang, C.-P. Green Chem. 2018, 20, 1662.
(f) Yang, J.; Zhao, H.-W.; He, J.; Zhang, C.-P. Catalysts 2018, 8, 23.
(g) Barata-Vallejo, S.; Cooke, M. V.; Postigo, A. ACS Catal. 2018, 8, 7287.
(a) Han, J.-B.; Hao, J.-H.; Zhang, C.-P.; Qin, H.-L. Curr. Org. Chem. 2015, 19, 1554 and the references cited therein.
(b) Li, L.; Huang, M.; Liu, C.; Xiao, J.-C.; Chen, Q.-Y.; Guo, Y.; Zhao, Z.-G. Org. Lett. 2015, 17, 4714.
(c) Tang, X.-J.; Thomoson, C. S.; Dolbier, Jr. W. R. Org. Lett. 2014, 16, 4594.
(d) Han, E.-J.; Sun, Y.; Shen, Q.; Chen, Q.-Y.; Guo, Y.; Huang, Y.-G. Org. Chem. Front. 2015, 2, 1379.
(e) Luo, H.; Wu, G.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2015, 54, 14503.
(f) Yu, X.; Cohen, S. M. J. Am. Chem. Soc. 2016, 138, 12320.
(g) Mai, W.-P.; Sun, B.; Qian, G.-S.; Yuan, J.-W.; Mao, P.; Yang, L.-R.; Xiao, Y.-M. Tetrahedron 2015, 71, 8416.
(h) Roh, G.-b.; Iqbal, N.; Cho, E. J. Chin. J. Chem. 2016, 34, 459.
(i) Zhang, Y.; Du, H.; Zhu, M.; Li, J.; Zou, D.; Wu, Y.; Wu, Y. Tetrahedron Lett. 2017, 58, 880.
(j) Zheng, J.; Chen, Q.-Y.; Sun, K.; Huang, Y.; Guo, Y. Tetrahedron Lett. 2016, 57, 5757.
(k) Andrews, K. G.; Faizova, R.; Denton, R. M. Nat. Commun. 2017, 8, 15913.
(a) Umemoto, T.; Gotoh, Y. J. Fluorine Chem. 1985, 28, 235.
(b) Umemoto, T.; Gotoh, Y. Bull. Chem. Soc. Jpn. 1987, 60, 3307.
(c) Lyalin, V. V.; Orda, V. V.; Alekseeva, L. A.; Yagupol'skii, L. M. J. Org. Chem. USSR 1972, 8, 1027.
(a) Zhdankin, V. V.; Kuehl, C. K.; Simonsen, A. J. Tetrahedron Lett. 1995, 36, 2203.
(b) Zhdankin, V. V.; Kuehl, C. K.; Simonsen, A. J. J. Org. Chem. 1996, 61, 8272.
Montanari, V.; Resnati, G. Tetrahedron Lett. 1994, 35, 8015. doi: 10.1016/0040-4039(94)80037-5
DesMarteau, D. D.; Montanari, V. Chem. Commun. 1998, 2241. https://pubs.rsc.org/en/Content/ArticleLanding/CC/1998/A805879B#!divAbstract
Umemoto, T; Gotoh, Y. J. Fluorine Chem. 1986, 31, 231. doi: 10.1016/S0022-1139(00)80536-9
Umemoto, T.; Gotoh, Y. Bull. Chem. Soc. Jpn. 1991, 64, 2008 doi: 10.1246/bcsj.64.2008
Tolnai, G. L.; Nilsson, U. J.; Olofsson, B. Angew. Chem., Int. Ed. 2016, 55, 11226. doi: 10.1002/anie.201605999
Han, Q.-Y.; Zhao, C.-L.; Yang, J.; Zhang, C.-P. Green Chem. Lett. Rev. 2017, 10, 162. doi: 10.1080/17518253.2017.1338759
Umemoto, T; Gotoh, Y. Bull. Chem. Soc. Jpn. 1987, 60, 3823. doi: 10.1246/bcsj.60.3823
Zhao, C.-L.; Yang, J.; Han, Z.-Z.; Zhang, C.-P. J. Fluorine Chem. 2017, 204, 23. doi: 10.1016/j.jfluchem.2017.09.009
Tolnai, G. L.; Székely, A.; Makó, Z.; Gáti, T.; Daru, J.; Bihari, T.; Stirling, A.; Novák, Z. Chem. Commun. 2105, 51, 4488.
DesMarteau, D. D.; Montanari, V. Chem. Lett. 2000, 29, 1052. doi: 10.1246/cl.2000.1052
(a) DesMarteau, D. D.; Lu, C. Tetrahedron Lett. 2006, 47, 561.
(b) DesMarteau, D. D.; Lu, C. J. Fluorine Chem. 2007, 128, 1326.
(a) Lu, C.; DesMarteau, D. D. J. Fluorine Chem. 2007, 128, 832. (b) Lu, C.; DesMarteau, D. D. Chem. Commun. 2008, 208.
(a) Zhang, J.; Martin, G. R.; DesMarteau, D. D. Chem. Commun. 2003, 2334.
(b) Lu, C.; VanDerveer, D.; DesMarteau, D. D. Org. Lett. 2008, 10, 5565.
Chu, A-H. A.; Minciunescu, A.; Montanari, V.; Kumar, K.; Bennett, S. C. Org. Lett. 2014, 16, 1780. doi: 10.1021/ol5004059
Chu, A-H. A.; Minciunescu, A.; Bennett, S. C. Org. Lett. 2015, 17, 6262. doi: 10.1021/acs.orglett.5b03282
(a) Yan, S.-Y.; Zhang, Z.-Z.; Shi, B.-F. Chem. Commun. 2017, 53, 10287.
(b) Zhang, H.; Wang, H.-Y.; Luo, Y.; Chen, C.; Cao, Y.; Chen, P.; Guo, Y.-L.; Lan, Y.; Liu, G. ACS Catal. 2018, 8, 2173.
(c) Zhang, X.; Yang, C. Adv. Synth. Catal. 2015, 357, 2721.
(d) Ohtsuka, Y.; Yamakawa, T. J. Fluorine Chem. 2016, 185, 96.
(e) Zhu, M.; Han, X.; Fu, W.; Wang, Z.; Ji, B.; Hao, X.-Q.; Song, M.-P.; Xu, C. J. Org. Chem. 2016, 81, 7282.
Yang, J.; Han, Q.-Y.; Zhao, C.-L.; Dong, T.; Hou, Z.-Y.; Qin, H.-L.; Zhang, C.-P. Org. Biomol. Chem. 2016, 14, 7654. doi: 10.1039/C6OB01384H
Tóth, B. L.; Kovács, S.; Sályi, G.; Novák, Z. Angew. Chem., Int. Ed. 2016, 55, 1988. doi: 10.1002/anie.201510555
Kovács, S.; Tóth, B. L.; Borsik, G.; Borsik, G.; Bihari, T.; May, N. V.; Stirling, A.; Novák, Z. Adv. Synth. Catal. 2017, 359, 527. doi: 10.1002/adsc.201601136
Borah, A. J.; Shi, Z. Chem. Commun. 2017, 53, 3945. doi: 10.1039/C7CC01274H
Maraswami, M.; Pankajakshan, S.; Chen, G.; Loh, T. P. Org. Lett. 2017, 19, 4223. doi: 10.1021/acs.orglett.7b01859
Wen, D.; Yuan, B.; He, R.; Shen, W.; Li, M. Tetrahedron Lett. 2018, 59, 462. doi: 10.1016/j.tetlet.2017.12.075

Scheme 4 Synthesis of phenyl(trifluoroethyl)iodonium bis(trifluoromethanesulfonyl)amide from CF3CH2I

Scheme 5 Electrophilic trifluoroethylation of different types of nucleophiles by [PhICH2CF3][OTf]

Scheme 10 Trifluoroethylation of enol silyl ethers with [PhICH2CF3][OTf] promoted by KF

Scheme 11 Structure-dependent selective O/C-trifluoroethylation of 1, 3-diketones with [MesCH2CF3][OTf]

Scheme 13 Trifluoroethylation of amino acids and peptides by [PhCH2CF3][NTf2] in aqueous media

Scheme 15 Synthesis of mono Nα-trifluoroethylated cyclic dipeptide from Nα-trifluoroethylated L-tyrosine

Scheme 16 Synthesis of Nα-trifluoroethyl L-phenylalanine methyl ester linear for construction of peptides containing a N-trifluoroethylated backbone amide linkage

Scheme 17 Trifluoroethylation of pyridine and imidazole derivatives with [PhICH2CF3][NTf2]

Scheme 18 [PhICH2CF3][NTf2]-promoted glycosylation of thioglycosides at room temperature

Scheme 19 A combination of [ArICH2CF3][NTf2] and nitrile solvent systems for stereoselective synthesis of armed 1, 2-trans-β-glycosides

Scheme 20 Pd-catalyzed divergent trifluoroethylation and arylation of arylboronic acids by [ArICH2CF3][OTf]

Scheme 23 Pd-Catalyzed trifluoroethylation of indoles at the C4 position with [MesICH2CF3][OTf]

Scheme 24 Pd-catalyzed trifluoroethylation of aromatic amides with [MesICH2CF3][OTf] via C—H activation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:











