
Scheme 1.
Nickel-catalyzed defluorinative coupling of gem-difluoroalkenes with lithium organoborate.

Due to similarity in spatial configuration and electrical distribution, monofluoroakene is often considered by medicinal chemists as a more lipophilic and metabolically more stable bioisostere of an amide in peptides [1-4], which was recently demonstrated to be a new type of powerful therapeutic agents for the treatment of human diseases [5-12]. Nevertheless, stereoselective preparation of (Z)- or (E)-monofluoroalkenes by classic Wittig reaction or Julia-Kocienski olefination was rather difficult, as reported previously [13-15]. Instead, an approach that utilized easily available gem-difluoroalkene as a proper precursor and an appropriate coupling partner under transition metal catalysis have emerged recently as an efficient method for the preparation of (Z)- or (E)-monofluoroalkenes in high stereoselectivity [4]. In this respect, several different types of commonly used coupling partners including Grignard reagents [16, 17], organozinc reagents [18], alkyl halide [19], silicon reagents [20], organoboron reagents [21, 22] have been successfully identified to be suitable for such a transformation to give defluorinative alkylation, silylation or boronization in high yields and excellent stereoselectivity. For instance, in 2015, Cao and coworkers reported a nickel-catalyzed highly Z-selective defluoroarylation of gem-difluoroalkenes with arylboronic acids [21]. Meanwhile, Toste and coworkers reported a palladium-catalyzed defluorinative coupling of gem-difluoroalkenes with aryl boronic acids, which was proposed to proceed through a β-fluoride elimination of a Pd(II) intermediate [22].
Very recently, during the development of nickel-catalyzed asymmetric cross-coupling reaction for the construction of fluoroalkyl-substituted stereogenic centers, we discovered that the combination of lithium organoborate with zinc bromide generated a zincate species which was more reactive toward transmetalating the aryl group from boron to nickel [23-26]. Inspired by such a phenomenon, we questioned ourselves whether such an in situ generated zincate species could be used in the coupling with gem-difluoroalkenes to stereoselectively generate monofluoroalkenes. Herein, we reported that in the presence of a nickel catalyst, coupling of gem-difluoroalkenes with a combination of various readily available "ate-type" aryl pinacol boronates and ZnBr2 occurred smoothly under mild conditions to give Z-monofluoroalkenes in good yields and excellent stereoselectivity (Scheme 1).
We chose the reaction of 2-(2, 2-difluorovinyl)naphthalene 1a with lithium organoborate, which was easily formed in situ by mixing phenylboronic acid pinacol ester with n-BuLi under −20 ℃ [27, 28], as the model reaction to optimize the reaction conditions. We initially discovered that in the presence of 5.0 mol% NiBr2•DME and 2.5 mol% of 4, 4′-dimethyl-2, 2′-bipyridine ligand L1, reaction of 2-(2, 2-difluorovinyl)naphthalene 1a with lithium phenylorganoborate 2a and 50 mol% of ZnBr2 in a number of common organic solvents, such as Et2O, THF, DME, DMF or toluene did not proceed into full conversion after 12 h at 0 ℃ and the yields for the desired to monofluoroalkene 3a were low (Table 1, entries 1–5). However, stereoselectivity of the reaction was excellent since only Z-monofluoroalkene 3a was observed in 19F NMR spectroscopies. To our delight, when a mixed solvent of Et2O/THF (9:1) was employed, the yield of compound 3a was significantly improved to 94% along the formation of single Z-isomer (Table 1, entry 6). Decreasing the ratio of Et2O in the mixed solvents from 9:1 to 4:1 has little effect on the yield and selectivity of the reaction. Yet, further decrease the ratio of Et2O in the mixed solvents to 1:1 led to significantly decrease the yield of the reaction to 80% (Table 1, entries 7 and 8). The choice of different nickel catalyst precursors and ligands played an essential role for the high yielding formation of compound 3a. When NiI2 was used as the catalyst precursor, the yield decreased dramatically (Table 1, entry 9). On the other hand, reactions using NiCl2•DME or Ni(OAc)2 as the catalyst gave the coupled products in slightly lower yields (Table 1, entries 10 and 11). In contrast, when a Ni(0) complex Ni(cod)2 was used as the precursor, the yield of the reaction deceased significantly to 48% (Table 1, entry 12). Likewise, the electronic properties of the ligand were also equally important for the high yields of the reaction. Using either more electron-donating or electron-poor bipyridine derivatives resulted in lower yields (Table 1, entries 13–16). Further optimization disclosed that the reaction conducted at room temperature gave lower yield (Table 1, entry 17). In addition, it was found that reaction using ZnI2 as an additive gave the defluoroarylation product 3a in slightly low yields, while reactions using other additives such as Zn(OTf)2 or MgBr2 were not effective at all (Table 1, entries 18–20). Our initially studies showed that the amount of ZnBr2 was important for the conversion of the reaction. Nevertheless, it was found that the yields decreased with the increase of the amount of ZnBr2 to 0.8 or 1.5 equiv. The effect of the addition of ZnBr2 to a solution of lithium aryl borate has been reported to generate lithium aryl zincates [PhxZnBry]n− (n = x + y − 2) [29], which facilitates the transmetalation step of the nickel-catalyzed cross coupling reaction [24]. Alternatively, ZnBr2 might act as a Lewis acid to abstract the bromide on the nickel metal center to facilitate the transmetalation step. Finally, control experiments showed that reactions in the absence of either nickel precursor, the ligand or additive ZnBr2 occurred in less than 10% yield (Table 1, entries 21–24).
 DownLoad:
CSV
DownLoad:
CSV

|
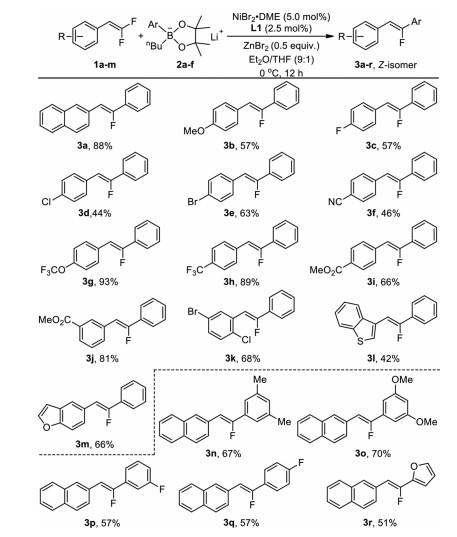
Having identified the suitable conditions for highly stereoselective formation of Z-monofluoroalkene 3a, we next examined the generality of the nickel-catalyzed defluorinative coupling of diverse gem-difluoroalkenes with a variety of aryl lithium organoborates under the optimized reaction conditions as shown in entry 6 in Table 1. As summarized in Scheme 1, in general, gem-difluoroalkenes with electron-donating or electron-withdrawing substituents at ortho-, meta- or para-position of the aryl moiety reacted smoothly to give the corresponding Z-monofluoroalkenes 3a-k in medium to good yields. For instance, reactions of lithium phenyl n-butyl borate 2a with gem-difluoroalkenes bearing an electron-donating para-methoxyl group occurred to give compound 3b in 57% yield, while reaction with gem-difluoroalkenes bearing an electron-withdrawing para-cyanophenyl group occurred to give compound 3f in 46% yield (Scheme 2, 3b and 3f). gem-Difluoroalkenes with a heteroaryl substituent such as benzothiophene or benzofuran also reacted smoothly to afford the corresponding Z-monofluoroalkenes 3l and 3m in 42% and 66% yields, respectively (Scheme 2, 3l and 3m). On the other hand, aryl boronic acid pinacol esters bearing methyl, methoxyl, fluoro and furan also reacted with gem-difluoroalkenes 1a to afford the defluorinative coupling products 3n-3r in 51%−70% yields (Scheme 2, 3n-3r). Because of the benign nature of the combination of lithium aryl borates and ZnBr2, a variety of common functional groups such as fluoride (3c, 3p, 3q), chloride (3d, 3k), bromide (3e, 3k), cyano (3f) and ester (3i, 3j) were compatible, which would allow for further functional group transformation.
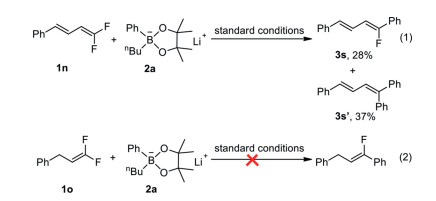
To further expand the scope of the reaction, we also explored other gem-difluoroalkenes including conjugated gem-difluoroalkene 1n and alkyl gem-difluoroalkene 1o (Scheme 3). We found that conjugated gem-difluoroalkene 1n reacted under the optimized conditions to give compound 3s in 28% yield, along with a secondary defluoroarylative product 3s' in 37% yield, while gem-difluoroalkene 1o with an alkyl group did not react under the optimal conditions at all.
As we mentioned in the introduction, monofluoroalkene is considered to be bioisostere of amide bond that is widespread presence in a number of bioactive compounds with diverse pharmacological activities, such as anti-cancer [30, 31], anti-diabetic [32, 33]. In 2016, Liu and coworkers reported that compound 4 was a more selective c-KIT inhibitor than Imatinib and Sunitinib for gastrointestinal stromal tumors (GISTs), which completely abolished ABL and FLT3 kinase [34]. To demonstrate the applicability of the current protocol, we synthesized a Z-monofluoroalkene mimic of the core structure of compound 4, as shown in Scheme 4. Treatment of 4-(2, 2-difluorovinyl)-2-(methoxymethoxy)-1-methylbenzene 1o with lithium m-trifluoromethylphenyl borate under standard conditions occurred smoothly to give corresponding product 3t in 95% yield (Scheme 4). Notably, this gram-scale reaction afforded the desired defluoroarylated product in pure Z-isomer.

In summary, a highly Z-selective construction of monofluoroalkenes via a nickel-catalyzed coupling of lithium organoborate with gem-difluoroalkenes was reported. In this reaction, the presence of ZnBr2 as an additive was crucial to achieve high reactivity and selectivity. The absence of base to activate organoboron and mild conditions allow the tolerance among a variety of useful functional groups in this coupling reaction. The applicability of the current method was demonstrated in the gram-scale synthesis of the monofluoroalkene mimic of the core structure of drug molecule compound 4. Expanding the scope of the reaction with fluorinated arenes and heteroarenes are currently undergoing in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors gratefully acknowledge the National Natural Science Foundation of China (Nos. 21625206, 21632009, 21421002, 22061160465) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi: 10.1016/j.cclet.2022.01.020.
K. Zhao, D. Lim, T. Funaki, J.T. Welch, Bioorg. Med. Chem. Lett. 11 (2003) 207–215. doi: 10.1016/S0968-0896(02)00384-X
P. Van der Veken, K. Senten, I. Kertesz, et al., J. Med. Chem. 48 (2005) 1768–1780. doi: 10.1021/jm0495982
N. Asakura, Y. Usuki, H. Iio, T. Tanaka, J. Fluorine Chem. 127 (2006) 800–808. doi: 10.1016/j.jfluchem.2006.02.016
J.J. Urban, B.G. Tillman, W.A. Cronin, J. Phys. Chem. A 110 (2006) 11120–11129.
S. Couve-Bonnaire, D. Cahard, X. Pannecoucke, Org. Biomol. Chem. 5 (2007) 1151–1157. doi: 10.1039/b701559c
B. Zajc, R. Kumar, Synthesis 2010 (2010) 1822-1836. doi: 10.1055/s-0029-1218789
H.J. Tsai, Tetrahedron Lett. 37 (1996) 629–632. doi: 10.1016/0040-4039(95)02218-X
H. Yanai, T. Taguchi, Eur. J. Org. Chem. (2011) 5939–5954 2011.
G. Landelle, M. Bergeron, M.O. Turcotte-Savard, J.F. Paquin, Chem. Soc. Rev. 40 (2011) 2867–2908. doi: 10.1039/c0cs00201a
A.K. Ghosh, B. Zajc, Org. Lett. 8 (2006) 1553–1556. doi: 10.1021/ol060002+
B. Zajc, S. Kake, Org. Lett. 8 (2006) 4457–4460. doi: 10.1021/ol0616236
Y.C. Zhao, F.Z. Jiang, J.B. Hu, J. Am. Chem. Soc. 137 (2015) 5199–5203. doi: 10.1021/jacs.5b02112
F. Liao, J. Yu, J. Zhou, Chin. J. Org. Chem. 37 (2017) 2175–2186. doi: 10.6023/cjoc201705001
X. Zhang, S. Cao, Tetrahedron Lett. 58 (2017) 375–392. doi: 10.1016/j.tetlet.2016.12.054
J. Paquin, M. Drouin, J. Hamel, Synthesis (Mass) 50 (2018) 881–955. doi: 10.1055/s-0036-1591867
W. Dai, J. Xiao, G. Jin, et al., J. Org. Chem. 79 (2014) 10537–10546. doi: 10.1021/jo5022234
W. Dai, H. Shi, X. Zhao, S. Cao, Org. Lett. 18 (2016) 4284–4287. doi: 10.1021/acs.orglett.6b02026
J. Zhang, B. Wang, Y. Liu, S. Cao, Chin, J. Org. Chem. 39 (2019) 249–256.
X. Lu, Y. Wang, B. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 12632–12637. doi: 10.1021/jacs.7b06469
D. Tan, E. Lin, W.W. Ji, et al., Adv. Synth. Catal. 360 (2018) 1032–1037. doi: 10.1002/adsc.201701497
Y. Xiong, T. Huang, X. Ji, J. Wu, S. Cao, Org. Biomol. Chem. 13 (2015) 7389–7392. doi: 10.1039/C5OB01016K
R.T. Thornbury, F.D. Toste, Angew. Chem. Int. Ed. 55 (2016) 11629–11632. doi: 10.1002/anie.201605651
W.C. Huang, X. Wan, Q. Shen, Angew. Chem. Int. Ed. 56 (2017) 11986–11989. doi: 10.1002/anie.201706868
W.C. Huang, M. Hu, X.B. Leng, Q. Shen, Nat. Commun. 10 (2019) 2963. doi: 10.1038/s41467-019-10851-4
S.S. Liu, W.C. Huang, D.C. Wang, et al., Org. Chem. Front. 10 (2019) 2630–2634.
W.C. Huang, X.L. Wan, Q. Shen, Org. Lett. 22 (2020) 4327–4332. doi: 10.1021/acs.orglett.0c01363
Y. Kobayashi, R. Mizojiri, Tetrahedron Lett. 37 (1996) 8531–8534. doi: 10.1016/0040-4039(96)01984-3
Y. Kobayashi, Y. Nakayama, R. Mizojiri, Tetrahedron 54 (1998) 1053–1062. doi: 10.1016/S0040-4020(97)10206-X
R.J. Procter, J.J. Dunsford, P.J. Rushworth, et al., Chem. Eur. J. 23 (2017) 15889–15893. doi: 10.1002/chem.201704170
S. Osada, S. Sano, M. Ueyama, et al., Bioorg. Med. Chem. 18 (2010) 605–611. doi: 10.1016/j.bmc.2009.12.005
J. Kanazawa, T. Takehashi, S. Akinaga, et al., Anticancer Drugs 9 (1998) 653–657. doi: 10.1097/00001813-199808000-00011
T. Deng, S. Shan, Z. Li, et al., Biol. Pharm. Bull. 28 (2005) 1192–1196. doi: 10.1248/bpb.28.1192
S.D. Edmondson, L. Wei, J. Xu, et al., Bioorg. Med. Chem. Lett. 18 (2008) 2409–2413.
Q. Wang, F. Liu, B. Wang, et al., J. Med. Chem. 59 (2016) 3964–3979.

Scheme 1 Nickel-catalyzed defluorinative coupling of gem-difluoroalkenes with lithium organoborate.

Scheme 2 Nickel-catalyzed defluorinative coupling of gem-difluoroalkenes with lithium organoborates. Reaction conditions: gem-difluoroalkenes (0.3 mmol), boron compound (0.9 mmol), NiBr2•DME (5.0 mol%), L1 (2.5 mol%), ZnBr2 (0.5 equiv.) in Et2O/THF (9:1) at 0 ℃ for 12 h. Isolated yields.

Scheme 4 Application of nickel-catalyzed defluorinative coupling of gem-difluoroalkenes.
Table 1. Optimization of the reaction conditions for nickel-catalyzed defluorinative coupling of gem-difluoroalkenes.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
