Scheme 1.
Synthesis of sterically hindered amides
Synthesis of Sterically Hindered and Electron-Deficient Secondary Amides from Unactivated Carboxylic Acids and Isothiocyanates
Jiaxi Tan , Ye Guo , Fei Zeng , Guanrong Chen , Longyong Xie , Weimin He

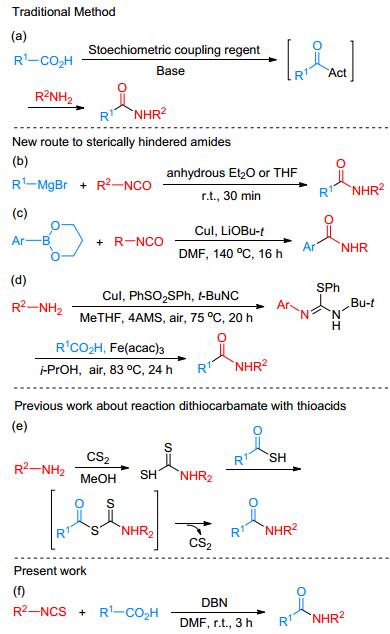
Amides are one of the most versatile and significant motifs in biologically natural products and synthetic drugs.[1] Therefore, the assembly of amides from the nature-abundant and low-cost starting materials has been an important objective in organic chemistry.[2] A number of synthetic strategies have been developed for their synthesis during the past decades.[3] Owing to the abundant and readily available carboxylic acids with ubiquitous functional groups, the construction of amides through dehydrative condensation reactions of carboxylic acids with amines has been dominantly employed in recent years (Scheme 1a).[2a~2c, 4] However, most current amide coupling reactions are still carried out in the presence of superstoichiometric amounts of expensive and often dangerous coupling reagents to facilitate the reaction, which leads to not only high-cost but also environmental issues.
Among the multifarious amides, sterically-hindered and electron-deficient amides are of particular interest, given diverse biological activities of these compounds.[5] However, to the best of our knowledge, only few examples of synthesizing sterically-hindered and electron-deficient amides have been reported. Bode et al.[6] first treated carboxylic acids with Grignard reagents to form sterically-hindered and electron-deficient secondary amides (Scheme 1b). However, the applicability and efficiency of this process is compromised by the use of air- and moisture sensitive Grignard reagent, which has limited commercial availability for alkyl- or aryl-variants as well as high cost. Zhang and coworkers[7] have pioneered the copper(I) catalyzed amidation reaction of organoboronic esters with isocyanates, but only arylamides were formed. Moreover, the difficult preparation step of the noncommercially available organoboronate ester, high temperature, long reaction time and inevitable residual metal in the final products have imposed limitations on the applicability of this method. Maes and coworkers[8] reported the Fe(acac)3-catalyzed amidation of carboxylic acids by using N-tert-butyl-S- methyl-N'-arylisothiourea as amino sources (Scheme 1d). However, the efficiency and atom economy of this protocol is compromised by the required pre-synthesis of N-tert-butyl-S-methyl-N'-arylisothiourea from moisture- sensitive reagents and unstable starting materials (tert- butyl isocyanide and S-phenyl benzenethiosulfonate)[9] that require tedious synthetic operations. In addition, the difficulties to remove the trace transition-metal contamination from the final products, particularly for the late-stage modification of medicine restrict their applications. It is therefore highly desired to develop an efficient and practical metal-free methodology that can achieve sterically-hin- dered and electron-deficient secondary amides using abundant and readily available materials under mild reaction conditions.[10]
In 2013, Yu and Houghten[11] first reported a traceless strategy for the synthesis of amides through coupling of thioacids and dithiocarbamates, the later of which served as the activated amino source. Although this modified process is found to be an improvement over previous methods, the inaccessible raw materials have prompted our to develop a straightforward approach for amides synthesis. Given that the isothiocyanate backbone is analogous to dithiocarbamate, we envisioned that the cheap and commercially available isothiocyanates could be used instead of dithiocarbamates. In light of the unique pharmacological activities of sterically-hindered and electron-deficient amides, and also in continuation of our efforts in mild organic synthesis, [12] herein, we report a metal-free, base-mediated amidation of carboxylic acids and isothiocyanates at ambient temperature (Scheme 1f). This approach offers a valuable alternative compared to the above-mentioned methods: (a) abundant and readily available raw materials are employed instead of air- and moisture sensitive Grignard reagents, expensive organoboronate esters and inaccessible N-tert-butyl-S-methyl-N'-arylisothiourea; (b) a broad range of amides, which include sterically-encumbered and electron-deficient secondary amides were formed under mild reaction reactions.
Our preliminary investigation focused on the reaction between benzoic acid 1a and phenyl isothiocyanate 2a with Et3N as the promoter in N, N-dimethylformamide (DMF) at room temperature, which resulted in 11% yield of N-phenylbenzamide 3aa based on 13% conversion of the 2a (Table 1, Entry 1). Fortunately, the first breakthrough was achieved when we replaced Et3N with 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base (Entry 2). Next, various bases were examined (Entries 3~6, 8), and 1, 5-diazabicyclo[4.3.0]non-5-ene (DBN) was found to be the most efficient base for this reaction (Entry 3). The investigation on the optimal amount of DBN (Entries 7, 9~8, 11) indicated that 2 equiv. DBN (Entries 7, 9) was an appropriate amount. The amidation did not take place without a base (Entries 9, 12). Further screening of the solvents showed that DMF was the best choice among those tested solvents (Entries 7, 9 vs 10, 13~16, 18). No improvement in the transformation was observed when ultrasonic radiation or microwave radiation was used instead of conventional heating (Entires 19, 20).
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Entry | Catalyst (equiv.) | Solvent | Yieldb/% |
| 1 | Et3N (1.5) | DMF | 11 |
| 2 | DBU (1.5) | DMF | 68 |
| 3 | DBN (1.5) | DMF | 90 |
| 4 | i-Pr2NEt (1.5) | DMF | Trace |
| 5 | Na2CO3 (1.5) | DMF | 6 |
| 6 | Cs2CO3 (1.5) | DMF | 9 |
| 7 | KF-Celite (1.5) | DMF | Trace |
| 8c | Amberlite IRA-4200 (10 wt%) | DMF | N.R. |
| 9 | DBN (2) | DMF | 97 |
| 10 | DBN (1.2) | DMF | 78 |
| 11 | DBN (0.2) | DMF | 12 |
| 12 | — | DMF | N.R. |
| 13 | DBN (2) | DCM | 8 |
| 14 | DBN (2) | THF | 15 |
| 15 | DBN (2) | MeCN | 72 |
| 16 | DBN (2) | Toluene | Trace |
| 17 | DBN (2) | DMSO | Trace |
| 18 | DBN (2) | MeOH | Trace |
| 19d | DBN (2) | DMF | 91 |
| 20e | DBN (2) | DMF | 94 |
| a Unless otherwise specified, the reactions were carried out in a vial in the presence of 1a (0.15 mmol), 2a (0.1 mmol), base, solvent (1 mL). b Estimated by 1H NMR spectroscopy using diethyl phthalate as an internal reference. c The quantity of Amberlite IRA-4200 was 0.1 g. d The reaction was conducted under 40 kHz/30 W ultrasonic radiation for 1 h. e The reaction was conducted under 150 W microwave radiation of 20 min. N.R.: no reaction. | |||
With the optimal system in hand, we examined the scope of carboxylic acid substrates with phenyl isothiocyanate 2a as illustrated in Table 2. To our delight, the current reaction system was suitable for a wide range of aliphatic carboxylic acids (3ba~3pa). Carboxylic acids with various chain lengths and isomeric structures did not significantly affect the product yields. High functional group compatibility was exhibited, such as tolerating alkyl (3ba~3ea), chlorine (3fa), acetyl (3ga), alkynyl (3ha), trifluoromethyl (3ia) and alkenyl (3ja) moieties. Remarkably, the steric effect of the substituted formic acids did not affect the yield of the reaction. For instance, carboxylic acids bearing bulky groups, such as iso-propyl-(3ka), tert-butyl (3la), cyclopropyl (3ma), 1-phenylcyclopropyl (3na), cyclohexyl (3oa) and 1-adamantyl (3pa), could be successfully transformed to the corresponding products in excellent yields. No matter whether the benzene ring of aryl carboxylic acid is substituted with either sterically hindered, electron-donating or electron-withdrawing group, all of them delivered the desired products in good to excellent yields (3aa, 3qa~3va). Polycyclic and heteroaromatic substituted carboxylic acids could also be transformed into the corresponding products in good to excellent yields (3wa~3ya). Interestingly, the reaction of the ferrocenecarboxylic acid gave the corresponding amide in good yield (3za), which highlighted the mild conditions of this reaction and potential applications of it in organocmetallic synthesis.
下载:
导出CSV
 |
 |
| a All reactions were carried out in a vail in the presence of 1 (0.45 mmol), 2 (0.3 mmol), DBN (0.6 mmol) and DMF (3 mL); isolated yields are reported. |
We next turned our attention to the scope of isothiocyanate substrates (Table 3). The amidation reaction was compatible with various sensitive functional groups, such as F-, Cl-, Br-, CF3-, CN- and NO2-substituted isothiocyanates. For instance, electron-donating groups, such as Me-, tert-Bu-, and MeO-promoted this transformation smoothly, yielding the desired amide products 3cb~3cd in excellent yields. Phenyl isothiocyanates with halogen groups (F, Cl, Br and I) are also suitable reaction partners (3ce~3ch). Less-reactive electron-withdrawing groups, such as CF3-, CN- and NO2-substituted isothiocyanates 1i~1k also successfully participated in the current reaction. Switching to meta-substituted phenyl isothiocyanates from para-substi- tuted substrates did not affect the outcome of the reaction (3cl~3cm), and sterically demanding ortho-substituted phenyl isothiocyanate also produced the expected product in moderate yields (3cn). 3, 5-Disubstituted phenyl isothiocyanate containing strong electron- withdrawing functionalities was also sucessfully applied to the reaction (3co). However, heteroaromatic and aliphatic isothiocyanates were ineffective, and only a trace amount of the desired products were detected.
下载:
导出CSV
 |
 |
| a All reactions were carried out in a vail in the presence of 1 (0.45 mmol), 2 (0.3 mmol), DBN (0.6 mmol) and DMF (3 mL); isolated yields are reported. |
Amides are not only valuable building blocks in synthetic chemistry but also important structure motifs in numerous biologically and pharmacologically active compounds. Moreover, late-stage structural modification is a highly valuable strategy for drug research and development. Therefore, several complex natural product derivatives were submitted to the standard reaction conditions (Table 4). Biologically active carboxylic acid derivatives, such as dehydroabietic acid, mycophenolic acid, norbornene acid and isoxepac, worked well in the current transformation, generating the corresponding amides in excellent yields (4ca~4cd).
下载:
导出CSV
 |
| a All reactions were carried out in a vail in the presence of 1 (0.45 mmol), 2 (0.3 mmol), DBN (0.6 mmol) and DMF (3 mL); isolated yields are reported. |
For the small-scale reaction, 1.5 equiv. of benzoic acid 1a were required, and the decrease in 1a loading would reduce the yield. Delightedly, the use of 1.3 equiv. of 1a could afford the desired product in 91% yield (1.07 g) with increase of the scale of the reaction by 20-fold (Eq. 1), demonstrating the synthetic utility of this protocol from a practical point of view.
|
|
(2) |
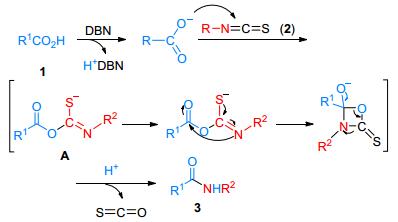
The amidation reaction may follow a similar mechanism to that of the reaction of thioacids and dithiocarbamateas reported by Yu and Houghten, with the major difference being isothiocyanates as the amino source instead of dithiocarbamate and carboxylic acids instead of thioacid.[11] First, the abstraction of proton from carboxylic acid by DBN generates the carboxylate anion, which attacks the sp-carbon of isothiocyanate 2 to yield the intermediate A. The intermediate A then proceeds via an intramolecular rearrangement reaction to afford product 3 (Scheme 2).
In summary, we have presented a simple and direct C(O)—N coupling method for the efficient preparation of amides starting with readily available unactivated carboxylic acid and isothiocyanates using common and cheap DBN as a mild base. Reactions occur readily at ambient temperature with good functional-group tolerance and can be easily scaled-up. Importantly, these transformations occur without the help of any transition metal, and a wide array of amides can be readily obtained in good to excellent yields. This protocol provides an alternative route to access sterically hindered and electron-deficient secondary amides which are difficult to synthesize through a conventional carboxyl-amine coupling reaction. This new method would be of significant use in the amide preparation for pharmaceuticals, natural products, and functional materials.
In a vial was placed carboxylic acids 1 (0.45 mmol), DMF (3 mL), DBN (0.6 mmol, 74 mg) and isothiocyanate 2 (0.3 mmol), then the contents were stirred at room temperature. After completion of the reaction (monitored by TLC), the reaction was quenched with water (5 mL), extracted with CH2Cl2 (5 mL×3), and the organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (eluent: hexanes/ethyl acetate) to afford 3.
Method A (purify by column chromatography): In a round-bottom flask was placed benzoic acid 1a (7.8 mol, 0.73 g), DMF (60 mL), DBN (12 mmol, 1.49 g) and phenyl isothiocyanate (2a) (6 mmol, 0.81 g), then the contents were stirred at room temperature. After completion of the reaction (monitored by TLC), the reaction was quenched with water (5mL), extracted with CH2Cl2 (30 mL×3), and the organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (eluent: hexane/ethyl acetate) to afford 1.07 g of 3aa in 91% yield.
Method B (purify by recrystallization): In a round-bot- tom flask was placed benzoic acid (1a) (7.8 mol, 0.73 g), DMF (60 mL), DBN (12 mmol, 1.49 g) and phenyl isothiocyanate (2a) (6 mmol, 0.81 g), then the contents were stirred at room temperature. After completion of the reaction (monitored by TLC), the reaction was quenched with water (5ml), extracted with CH2Cl2 (30 mL×3), and the organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by recrystallization with a mixed solvent of ethyl acetate/petrol ether (30 mL, V:V=1:8) to give 0.99 g of 3aa in 84% yield.
N-Phenylbenzamide (3aa):[131] H NMR (400 MHz, DMSO-d6) δ: 10.27 (s, 1H), 7.96~7.94 (m, 2H), 7.78 (d, J=8.4 Hz, 2H), 7.60~7.51 (m, 3H), 7.35 (t, J=8.4 Hz, 2H), 7.10 (t, J=7.2 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 168.1, 139.7, 135.5, 132.1, 129.2, 128.9, 128.2, 124.2, 120.9.
N-Phenylacetamide (3ba):[14]1H NMR (400 MHz, DMSO-d6) δ: 9.93 (s, 1H), 7.57 (d, J=7.6 Hz, 2H), 7.28 (t, J=7.6 Hz, 2H), 7.01 (t, J=7.6 Hz, 1H), 2.04 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.4, 139.5, 128.8, 123.1, 119.1, 24.1.
N-Phenylpropionamide (3ca):[15]1H NMR (400 MHz, DMSO-d6) δ: 9.86 (s, 1H), 7.59 (d, J=8.4 Hz, 2H), 7.28 (t, J=7.6 Hz, 2H), 7.01(t, J=6.8 Hz, 1H), 2.34~2.81 (m, 2H), 1.07 (t, J=7.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 172.5, 139.9, 129.2, 123.4, 119.5, 30.0, 10.2.
N-Phenyloctanamide (3da):[16]1H NMR (400 MHz, DMSO-d6) δ: 9.86 (s, 1H), 7.59 (d, J=7.6 Hz, 2H), 7.27 (t, J=7.2 Hz, 2H), 7.00 (t, J=7.2 Hz, 1H), 2.82 (t, J=7.2 Hz, 2H), 1, 57 (s, 2H), 1.26 (d, J=9.6 Hz, 8H), 0.87~0.84 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 171.8, 139.9, 129.1, 123.4, 119.5, 36.9, 31.7, 29.1, 29.0, 25.7, 22.6, 14.5.
N, 3-Diphenylpropanamide (3ea):[17]1H NMR (400 MHz, DMSO-d6) δ: 9.93 (s, 1H), 7.60~7.58 (m, 2H), 7.30~7.26 (m, 6H), 7.20~7.16 (m, 1H), 7.02 (t, J=7.6 Hz, 1H), 2.91 (d, J=8.0 Hz, 2H), 2.65~2.61 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 170.9, 141.7, 139.8, 129.1, 128.9, 128.8, 126.5, 123.5, 119.6, 38.5, 31.4.
5-Chloro-N-phenylpentanamide (3fa): 1H NMR (400 MHz, DMSO-d6) δ: 9.90 (s, 1H), 7.58 (d, J=7.6 Hz, 2H), 7.28 (t, J=7.6 Hz, 2H), 7.02 (t, J=7.6 Hz, 1H), 3.66 (t, J=6.4 Hz, 2H), 2.34 (t, J=7.2 Hz, 2H), 1.78~1.68 (m, 4 H); 13C NMR (100 MHz, DMSO-d6) δ: 171.0, 139.4, 128.8, 123.1, 119.1, 45.2, 35.5, 31.7, 22.6; IR (KBr) ν: 3302, 2946, 1684, 1600, 1500, 1442, 754 cm-1; HRMS (ESI) calcd for C11H15ClNO [M+H]+ 212.0837, found 212.0833.
6-oxo-N-Phenylheptanamide (3ga): 1H NMR (400 MHz, DMSO-d6) δ: 9.94 (s, 1H), 7.64 (d, J=7.8 Hz, 2H), 7.34 (t, J=7.8 Hz, 2H), 7.07 (t, J=7.8 Hz, 1H), 2.52 (t, J=7.8 Hz, 2H), 2.35 (t, J=7.2 Hz, 2H), 2.13 (s, 3H), 1.64~1.50 (m, 4 H); 13C NMR (100 MHz, DMSO-d6) δ: 208.9, 171.6, 139.8, 129.2, 123.5, 119.5, 43.0, 36.8, 30.2, 25.2, 23.4; IR (KBr) ν: 3307, 2942, 1682, 1598, 1500, 1441, 755 cm-1; HRMS (ESI) calcd for C13H18NO2 [M+H]+ 220.1332, found 220.1329.
N-Phenylhept-6-ynamide (3ha): 1H NMR (400 MHz, DMSO-d6) δ: 9.89 (s, 1H), 7.59 (d, J=8.8 Hz, 2H), 7.28 (t, J=8.4 Hz, 2H), 7.01 (t, J=6.4 Hz, 1H), 2.76 (t, J=2.8 Hz, 1H), 2.31 ((t, J=7.6 Hz, 2H), 2.21~2.17 (m, 2H), 1.71~1.64 (m, 2H), 1.52~1.44 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 171.6, 139.8, 129.2, 123.5, 119.6, 84.9, 71.8, 36.3, 28.1, 24.8, 18.0; IR (KBr) ν: 3310, 2114, 1662, 1600, 1496, 1441, 754 cm-1; HRMS (ESI) calcd for C13H16NO [M+H]+ 202.1226, found 202.1230.
4, 4, 4-Trifluoro-N-phenylbutanamide (3ia): 1H NMR (400 MHz, DMSO-d6) δ: 10.09 (s, 1H), 7.58 (d, J=8.0 Hz, 2H), 7.30 (t, J=7.6 Hz, 2H), 7.04 (t, J=7.6 Hz, 1H), 2.63~2.55 (m, 4 H); 13C NMR (100 MHz, DMSO-d6) δ: 168.9, 139.6, 129.3, 128.1 (q, J=274.8 Hz), 123.8, 119.6, 29.2 (q, J=2.9 Hz), 29.0 (q, J=28.4 Hz); 19F NMR (376 MHz, DMSO-d6) δ: -65.2; IR (KBr) ν: 3314, 2947, 1665, 1599, 1494, 1442, 755 cm-1; HRMS (ESI) [M+H]+ calcd for C10H11F3NO: 218.0787, found 218.0781.
N-Phenylmethacrylamide (3ja):[18]1H NMR (400 MHz, DMSO-d6) δ: 9.78 (s, 1H), 7.69~7.66 (m, 2H), 7.32~7.28 (m, 2H), 7.07 (t, J=7.2 Hz, 1H), 5.79 (s, 1H), 5.51 (t, J=1.2 Hz, 1H), 1.94 (d, J=0.8 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 167.3, 140.9, 139.6, 129.0, 124.0, 120.7, 120.4, 19.3.
N-Phenylisobutyramide (3ka):[19]1H NMR (400 MHz, DMSO-d6) δ: 9.74 (s, 1H), 7.52 (d, J=7.6 Hz, 2H), 7.19 (t, J=7.6 Hz, 2H), 6.93(t, J=7.6 Hz, 1H), 2.43~2.41 (m, 1H), 1.01 (d, J=6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6) δ: 175.7, 140.0, 129.1, 123.4, 119.6, 35.4, 20.0.
N-Phenylpivalamide (3la):[20]1H NMR (400 MHz, DMSO-d6) δ: 9.25 (s, 1H), 7.69 (d, J=8.0 Hz, 2H), 7.34 (t, J=8.0 Hz, 2H), 7.09 (t, J=7.2 Hz, 1H), 1.28 (s, 9H); 13C NMR (100 MHz, DMSO-d6) δ: 176.8, 139.8, 128.9, 123.6, 120.7, 39.6, 27.7.
N-Phenylcyclopropanecarboxamide (3ma):[21]1H NMR (400 MHz, DMSO-d6) δ: 10.09 (s, 1H), 7.55 (d, J=8.4 Hz, 2H), 7.22 (t, J=8.0 Hz, 2H), 6.97 (t, J=8.0 Hz, 1H), 1.78~1.72 (m, 1H), 0.83~0.79 (m, 2H), 0.76~0.71 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 172.1, 139.8, 128.9, 123.2, 119.4, 14.9, 7.5.
N, 1-Diphenylcyclopropanecarboxamide (3na): 1H NMR (400 MHz, DMSO-d6) δ: 9.04 (s, 1H), 7.55~7.52 (m, 2H), 7.41~7.34 (m, 4H), 7.30~7.24 (m, 3H), 7.04~7.01 (m, 1H), 1.46~1.43 (m, 2H), 1.12~1.10 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 171.5, 140.8, 139.4, 129.4, 129.1, 129.0, 127.6, 124.0, 120.6, 32.3, 15.3; IR (KBr) ν: 3312, 3050, 2942, 1664, 1597, 1498, 1440, 753 cm-1; HRMS (ESI) calcd for C16H16NO [M+H]+ 238.1226, found 238.1225.
N-Phenylcyclohexanecarboxamide (3oa):[22]1H NMR (400 MHz, DMSO-d6) δ: 9.80 (s, 1H), 7.60(d, J=7.2 Hz, 2H), 7.29~7.25(m, 2H), 7.00 (t, J=7.6 Hz, 1H), 2.34~2.29 (m, 1H), 1.80~1.73 (m, 4H), 1.45~1.15 (m, 6H); 13C NMR (100 MHz, DMSO-d6) δ: 174.8, 140.0, 129.1, 123.3, 119.5, 45.4, 29.7, 25.9, 25.8.
N-Phenyladamantane-1-carboxamide (3pa):[23]1H NMR (400 MHz, DMSO-d6) δ: 9.11 (s, 1H), 7.66 (t, J=1.6 Hz, 2H), 7.29~7.26 (m, 2H), 7.02 (t, J=7.6 Hz, 1H), 2.01~1.69 (m, 15H); 13C NMR (100 MHz, DMSO-d6) δ: 176.4, 139.9, 128.9, 123.6, 120.7, 41.4, 38.8, 36.5, 28.2.
4-Methyl-N-phenylbenzamide (3qa):[13]1H NMR (400 MHz, DMSO-d6) δ: 10.17 (s, 1H), 7.89~7.77 (m, 4H), 7.36~7.32 (m, 4H), 7.13 (d, J=7.2 Hz, 1H), 2.38 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 165.9, 142.1, 139.8, 132.6, 129.4, 129.1, 128.2, 124.1, 120.8, 21.5.
4-Methoxy-N-phenylbenzamide (3ra):[24]1H NMR (400 MHz, DMSO-d6) δ: 10.09 (s, 1H), 7.95 (d, J=8.8 Hz, 2H), 7.76 (d, J=8.0 Hz, 2H), 7.34 (t, J=8.0 Hz, 2H), 7.10~7.05 (m, 3H), 3.84 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 165.5, 162.4, 139.9, 130.1, 129.1, 127.4, 123.9, 120.8, 114.1, 55.9.
4-Bromo-N-phenylbenzamide (3sa):[25]1H NMR (400 MHz, DMSO-d6) δ: 10.33 (s, 1H), 7.91 (d, J=8.4 Hz, 2H), 7.77~7.74 (m, 4 H), 7.36 (t, J=7.6 Hz, 2H), 7.11 (t, J=7.2 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 165.1, 139.5, 134.5, 131.9, 130.3, 129.2, 125.9, 124.4, 120.9.
Methyl 4-(phenylcarbamoyl)benzoate (3ta):[26]1H NMR (400 MHz, DMSO-d6) δ: 10.45 (s, 1H), 8.11~8.06 (m, 4H), 7.78 (d, J=8.8 Hz 2H), 7.37 (t, J=8.4 Hz, 2H), 7.12 (t, J=8.4 Hz, 1H), 3.90 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 166.2, 165.2, 139.6, 139.4, 132.5, 129.7, 129.2, 128.6, 124.5, 120.9, 53.0.
3-Methyl-N-phenylbenzamide (3ua):[25]1H NMR (400 MHz, DMSO-d6) δ: 10.22 (s, 1H), 7.79~7.77 (m, 4H), 7.44~7.40 (m, 4H), 7.12~7.07 (m, 1H), 2.40 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 166.2, 139.7, 138.2, 135.5, 132.6, 129.1, 128.8, 128.6, 125.3, 124.1, 120.8, 21.5.
2-Methyl-N-phenylbenzamide (3va):[25]1H NMR (400 MHz, DMSO-d6) δ: 10.31 (s, 1H), 7.75 (d, J=8.0 Hz, 2H), 7.46~7.30 (m, 6 H), 7.09 (t, J=7.2 Hz, 1H), 2.38 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.4, 139.8, 137.8, 135.7, 131.0, 130.1, 129.2, 127.7, 126.2, 124.0, 120.1, 19.8.
N-Phenyl-2-naphthamide (3wa):[27]1H NMR (400 MHz, DMSO-d6) δ: 10.45 (s, 1H), 8.59 (s, 1H), 8.11~8.01 (m, 4H), 7.83 (dd, J=8.8 Hz, 1.2 Hz, 2H), 7.67~7.61 (m, 2H), 7.38 (t, J=7.2 Hz, 2H), 7.14~7.10 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 166.1, 139.8, 134.8, 132.8, 132.6, 129.5, 129.2, 128.6, 128.5, 128.4, 128.2, 127.4, 125.0, 124.2, 120.9.
N-Phenyl-2, 3-dihydrobenzo[b][1, 4]dioxine-2-carboxamide (3xa): 1H NMR (400 MHz, DMSO-d6) δ: 10.16 (s, 1H), 7.65 (d, J=7.6 Hz, 2H), 7.33 (t, J=7.6 Hz, 2H), 7.11~7.03 (m, 2H), 6.91~6.87 (m, 3H), 4.99~4.97 (m, 1H), 4.48-4.33 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 166.1, 143.5, 143.0, 138.7, 129.3, 124.6, 122.2, 122.1, 120.5, 117.9, 117.6, 73.2, 65.3; IR (KBr) ν: 3310, 2945, 1665, 1594, 1497, 1441, 754 cm-1; HRMS (ESI) [M+H]+calcd for C15H14NO3: 256.0968, found 256.0959.
N-Phenylpicolinamide (3ya):[25]1H NMR (400 MHz, DMSO-d6) δ: 10.65 (s, 1H), 8.75~8.71 (m, 1H), 8.18~8.15 (m, 1H), 8.09~8.05 (m, 1H), 7.93 (d, J=1.2 Hz, 1H), 7.91~7.90 (d, J=0.8 Hz, 1H), 7.69~7.66 (m, 1H), 7.38~7.34 (m, 2H), 7.14~7.10 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 163.0, 149.1, 149.0, 138.7, 129.2, 127.5, 124.5, 122.9, 120.8.
N-Phenylferrocenylamide (3za):[28]1H NMR (400 MHz, DMSO-d6) δ: 9.44 (s, 1H), 7.70 (s, 2H), 7.33 (t, J=7.6 Hz, 2H), 7.06 (t, J=7.2 Hz, 1H), 5.01 (t, J=2.0 Hz, 2H), 4.45 (t, J=2.0 Hz, 2H), 4.21 (t, J=5.2 Hz, 5H); 13C NMR (100 MHz, DMSO-d6) δ: 168.6, 139.7, 129.0, 123.6, 120.8, 76.9, 71.0, 70.0, 69.1.
N-(p-Tolyl)acetamide (3cb):[14]1H NMR (400 MHz, DMSO-d6) δ: 9.84 (s, 1H), 7.45 (d, J=8.4 Hz, 2H), 7.09 (d, J=8.4 Hz, 2H), 2.23 (s, 3H), 2.01 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.5, 137.4, 132.3, 129.5, 119.5, 24.5, 20.9.
N-(4-(tert-Butyl)phenyl)acetamide (3cc):[29]1H NMR (400 MHz, DMSO-d6) δ: 9.86 (s, 1H), 7.48 (d, J=8.8 Hz, 2H), 7.29 (d, J=8.8 Hz, 2H), 2.01(s, 3H), 1.25 (s, 9 H); 13C NMR (100 MHz, DMSO-d6) δ: 168.5, 145.7, 137.3, 125.8, 119.3, 34.5, 31.7, 24.4.
N-(4-Methoxyphenyl)acetamide (3cd):[30]1H NMR (400 MHz, DMSO-d6) δ: 9.78 (s, 1H), 7.47 (d, J=8.8 Hz, 2H), 6.85 (d, J=8.8 Hz, 2H), 3.70 (s, 3H), 1.99 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.3, 155.5, 133.0, 121.0, 114.3, 55.6, 24.3.
N-(4-Fluorophenyl)acetamide (3ce):[31]1H NMR (400 MHz, DMSO-d6) δ: 10.00 (s, 1H), 7.60~7.57(m, 2H), 7.13 (t, J=8.8 Hz, 2H), 2.03 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.7, 158.3 (J=237.7 Hz), 136.3 (J=2.2 Hz), 121.2 (J=8 Hz), 115.7 (J=21.9 Hz), 24.4; 19F NMR (376 MHz, DMSO-d6) δ: -119.8.
N-(4-Chlorophenyl)acetamide (3cf):[32]1H NMR (400 MHz, DMSO-d6) δ: 10.07 (s, 1H), 7.60 (d, J=8.8 Hz, 2H), 7.33 (d, J=8.8 Hz, 2H), 2.04 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.5, 138.4, 128.7, 126.5, 120.5, 24.1.
N-(4-Bromophenyl)acetamide (3cg):[33]1H NMR (400 MHz, DMSO-d6) δ: 10.07 (s, 1H), 7.55 (d, J=8.8 Hz, 2H), 7.46 (d, J=8.8 Hz, 2H), 2.03 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.6, 138.8, 131.6, 121.0, 114.6, 24.1.
N-(4-Iodophenyl)acetamide (3ch):[34]1H NMR (400 MHz, DMSO-d6) δ: 10.05 (s, 1H), 7.60 (d, J=8.8 Hz, 2H), 7.41 (d, J=8.8 Hz, 2H), 2.03 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 169.1, 139.7, 137.9, 121.7, 86.9, 24.6.
N-(4-(Trifluoromethyl)phenyl)acetamide (3ci):[35]1H NMR (400 MHz, DMSO-d6) δ: 10.31 (s, 1H), 7.78 (d, J=8.4 Hz, 2H), 7.33 (d, J=8.4 Hz, 2H), 2.08 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 169.5, 143.4, 125.8 (q, J=32.0 Hz), 124.0 (q, J=270 Hz) 126.3, 121.0, 24.5; 19F NMR (376 MHz, DMSO-d6) δ: -60.4.
N-(4-Cyanophenyl)acetamide (3cj):[36]1H NMR (400 MHz, DMSO-d6) δ: 10.38 (s, 1H), 7.75~7.74 (m, 4 H), 2.09 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 169.3, 143.6, 133.4, 119.2, 119.0, 104.8, 24.3.
N-(4-Nitrophenyl)acetamide (3ck):[31]1H NMR (400 MHz, DMSO-d6) δ: 10.56 (s, 1H), 8.19 (d, J=8.8 Hz, 2H), 7.80 (d, J=8.8 Hz, 2H), 2.11 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 169.9, 146.0, 142.5, 125.5, 119.0, 24.8.
N-(m-Tolyl)acetamide (3cl):[37]1H NMR (400 MHz, DMSO-d6) δ: 9.85 (s, 1H), 7.41 (s, 1H), 7.35 (d, J=8.0 Hz, 1H), 7.15 (t, J=7.6 Hz, 1H), 6.84 (d, J=7.6 Hz, 1H), 2.26 (s, 3H), 2.02 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.7, 139.8, 138.3, 129.0, 124.2, 120.0, 116.7, 24.5, 21.7
N-(3-Fluorophenyl)acetamide (3cm):[38]1H NMR (400 MHz, DMSO-d6) δ: 10.16 (s, 1H), 7.61~7.57 (m, 1H), 7.34~7.24 (m, 2H), 6.86~6.81 (m, 1H), 2.05 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 169.3, 162.7 (d, J=239.2 Hz), 141.6 (d, J=10.9 Hz), 130.9 (d, J=9.5 Hz), 115.2 (d, J=2.9 Hz), 110.0 (d, J=20.4 Hz), 106.3 (d, J=26.2 Hz), 24.6; 19F NMR (376 MHz, DMSO-d6) δ: -112.2.
N-(o-Tolyl)acetamide (3cn):[39]1H NMR (400 MHz, DMSO-d6) δ: 9.29 (s, 1H), 7.38 (d, J=7.6 Hz, 1H), 7.20~7.04 (m, 3H), 2.19 (s, 3H), 2.05 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 168.7, 137.0, 132.0, 130.8, 126.4, 125.5, 125.5, 23.8, 18.4.
N-(3, 5-Bis(trifluoromethyl)phenyl)acetamide (3co): 1H NMR (400 MHz, DMSO-d6) δ: 10.56 (s, 1H), 8.20 (s, 2H), 7.58 (s, 1H), 2.08 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 169.9, 141.7, 131.3 (q, J=32.1 Hz), 122.4 (q, J=270.6 Hz), 118.9 (d, J=3.7 Hz), 116.0, 24.5; 19F NMR (376 MHz, DMSO-d6) δ: -62.3; IR (KBr) ν: 3312, 2944, 1665, 1597, 1498, 1440, 753 cm-1; HRMS (ESI) calcd for C10H8F6NO [M+H]+ 272.0505, found 272.0512.
(1S, 4aS, 10aR)-7-Isopropyl-1, 4a-dimethyl-N-phenyl-1, 2, 3, 4, 4a, 9, 10, 10a-octahydrophenanthrene-1-carboxamide (4ca): 1H NMR (400 MHz, DMSO-d6) δ: 9.31 (s, 1H), 7.60 (d, J=8.4 Hz, 2H), 7.28 (t, J=8.0 Hz, 2H), 7.19 (d, J=8.0 Hz, 1H), 7.05~6.97 (m, 2H), 6.84 (d, J=1.6 Hz, 1H), 2.82~2.73 (m, 3H), 2.31~2.22 (m, 2H), 1.86~1.68 (m, 4 H), 1.58~1.52 (m, 2H), 1.25~1.23 (m, 4H), 1.17~1.14 (m, 9H); 13C NMR (100 MHz, DMSO-d6) δ: 177.2, 147.7, 145.7, 140.0, 134.9, 129.0, 127.0, 124.7, 124.3, 123.8, 121.2, 48.1, 44.9, 37.8, 37.4, 36.4, 33.5, 30.1, 25.6, 24.6, 24.5, 21.3, 19.1, 17.0; IR (KBr) ν: 3316, 2925, 1664, 1598, 1500, 1442, 752 cm-1; HRMS (ESI) calcd for C26H34NO [M+H]+ 376.2635, found 376.2624.
6-(4-Hydroxy-6-methoxy-7-methyl-3-oxo-1, 3-dihydroisobenzofuran-5-yl)-4-methyl-N-phenylhex-4-enamide (4cb): 1H NMR (400 MHz, DMSO-d6) δ: 9.84 (s, 1H), 9.35 (s, 1H), 7.54~7.52 (m, 2H), 7.23 (t, J=7.6 Hz, 2H), 6.98 (t, J=7.2 Hz, 1H), 3.65 (s, 3H), 3.49 (s, 3H), 3.31 (d, J=6.8 Hz, 2H), 2.38 (t, J=6.0 Hz, 2H), 2.26 (t, J=8.0 Hz, 2H), 2.03 (s, 3H), 1.79 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 171.4, 170.9, 163.1, 153.4, 146.3, 139.9, 134.4, 129.1, 123.4, 123.4, 123.0, 119.6, 116.5, 107.5, 69.2, 61.1, 35.8, 35.4, 23.0, 16.6, 11.4; IR (KBr) ν: 3318, 2945, 1662, 1598, 1498, 1443, 754 cm-1; HRMS (ESI) calcd for C23H26NO5 [M+H]+ 396.1805, found 396.1811.
N-Phenylbicyclo[2.2.1]hept-5-ene-2-carboxamide (4cc): 1H NMR (400 MHz, DMSO-d6) δ: 9.75 (s, 1H), 7.55 (t, J=7.2 Hz, 2H), 7.28~7.24 (m, 2H), 6.99 (t, J=7.6 Hz, 1H), 6.17~6.15 (m, 1H), 5.85~5.83 (m, 1H), 3.28 (s, 1H), 3.05~3.00 (m, 1H), 2.87 (s, 1H), 1.83~1.77 (m, 1H), 1.42~1.30 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 172.4, 140.1, 137.7, 132.2, 129.1, 123.2, 119.6, 50.1, 46.7, 44.7, 42.8, 28.8; IR (KBr) ν: 3320, 2927, 1660, 1598, 1500, 1442, 755 cm-1; HRMS (ESI) calcd for C14H16NO [M+H]+ 214.1226, found 214.1222.
2-(11-oxo-6, 11-Dihydrodibenzo[b, e]oxepin-2-yl)-N-phenylacetamide (4cd): 1H NMR (400 MHz, DMSO-d6) δ: 10.23 (s, 1H), 8.07 (d, J=2.4 Hz, 1H), 7.95 (s, 2H), 7.78 (d, J=8 Hz, 1H), 7.67~7.54 (m, 4 H), 7.30 (t, J=7.6 Hz, 2H), 7.05 (m, 2H), 5.28 (s, 2H), 3.68 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 190.3, 169.1, 160.0, 140.1, 139.3, 136.8, 136.1, 133.2, 131.6, 129.9, 129.3, 128.9, 128.8, 128.4, 124.7, 123.4, 120.8, 119.2, 72.9, 42.3; IR (KBr) ν: 3317, 2948, 1663, 1599, 1498, 1440, 753 cm-1; HRMS (ESI) calcd for C22H18NO3 [M+H]+ 344.1281, found 344.1278.
Supporting Information 1H NMR, 13C NMR and HRMS spectra of products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(a) Greenberg, A. ; Breneman, C. M. ; Liebman, J. F. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, New York 2000.
(b) Roughley, S. D. ; Jordan, A. M. J. Med. Chem. 2011, 54, 3451.
(c) Hong, Y. ; Dai, H. ; Ye, L. ; Zhong, S. ; Cao, X. ; Shi, Y. ; Li, C. ; Shi, J. ; Shi, L. Chin. J. Org. Chem. 2017, 37, 3006 (in Chinese).
(洪宇, 戴红, 叶林玉, 仲苏林, 曹雄飞, 石玉军, 李春建, 石健, 施磊, 有机化学, 2017, 37, 3006. )
(d) Gao, H. ; Zheng, X. ; Qi, Y. ; Wang, S. ; Wan, C. ; Rao, G. ; Mao, Z. Chin. J. Org. Chem. 2018, 38, 648 (in Chinese).
(高慧, 郑喜, 祁燕, 王斯, 万春平, 饶高雄, 毛泽伟, 有机化学, 2018, 38, 648. )
(e) Qiao, L. ; Wei, Y. ; Hao, S. Chin. J. Org. Chem. 2018, 38, 509 (in Chinese).
(乔丽丽, 魏艳, 郝双红, 有机化学, 2018, 38, 509. )
(a) Han, S. -Y. ; Kim, Y. -A. Tetrahedron 2004, 60, 2447.
(b) Montalbetti, C. A. G. N. ; Falque, V. Tetrahedron 2005, 61, 10827.
(c) Lundberg, H. ; Tinnis, F. ; Selander, N. ; Adolfsson, H. Chem. Soc. Rev. 2014, 43, 2714.
(d) El-Faham, A. ; Albericio, F. Chem. Rev. 2011, 111, 6557.
(e) de Figueiredo, R. M. ; Suppo, J. -S. ; Campagne, J. -M. Chem. Rev. 2016, 116, 12029.
(f) Yang, J. ; Zhao, J. Sci. China Chem. 2018, 61, 97.
(g) Dong, H. ; Hou, M. Chin. J. Org. Chem. 2017, 37, 267 (in Chinese).
(董浩, 侯梅芳, 有机化学, 2017, 37, 267. )
(h) Yu, X. ; Zhou, F. ; Chen, J. ; Xiao, W. Acta Chim. Sinica 2017, 75, 86 (in Chinese).
(余晓叶, 周帆, 陈加荣, 肖文精, 化学学报, 2017, 75, 86. )
(a) Sasaki, K. ; Crich, D. Org. Lett. 2011, 13, 2256.
(b) Pan, J. ; Devarie-Baez, N. O. ; Xian, M. Org. Lett. 2011, 13, 1092.
(c) Liu, Z. ; Zhang, J. ; Chen, S. ; Shi, E. ; Xu, Y. ; Wan, X. Angew. Chem., Int. Ed. 2012, 51, 3231.
(d) Sun, X. ; Wang, M. ; Li, P. ; Zhang, X. ; Wang, L. Green Chem. 2013, 15, 3289.
(e) Li, D. ; Wang, M. ; Liu, J. ; Zhao, Q. ; Wang, L. Chem. Commun. 2013, 49, 3640.
(f) Liu, J. ; Liu, Q. ; Yi, H. ; Qin, C. ; Bai, R. ; Qi, X. ; Lan, Y. ; Lei, A. Angew. Chem., Int. Ed. 2014, 53, 502.
(g) Tang, C. ; Jiao, N. Angew. Chem., Int. Ed. 2014, 53, 6528.
(h) Schäfer, G. ; Bode, J. W. Org. Lett. 2014, 16, 1526.
(i) Du, H. ; Ruan, Q. ; Qi, M. ; Han, W. J. Org. Chem. 2015, 80, 7816.
(j) Lim, D. S. W. ; Lew, T. T. S. ; Zhang, Y. Org. Lett. 2015, 17, 6054.
(k) Debnath, P. ; Baeten, M. ; Lefèvre, N. ; Daele, S. V. ; Maes, B. U. W. Adv. Synth. Catal. 2015, 357, 197.
(l) Ali, M. A. ; Yao, X. ; Sun, H. ; Lu, H. Org. Lett. 2015, 17, 1513.
(m) Han, W. ; Jin, F. ; Zhao, Q. ; Du, H. ; Yao, L. Synlett 2016, 27, 1854.
(n) Gu, L. ; Wang, W. ; Liu, J. ; Li, G. ; Yuan, M. Green Chem. 2016, 18, 2604.
(o) Kaishap, P. P. ; Sarma, B. ; Gogoi, S. Chem. Commun. 2016, 52, 9809.
(p) Ren, L. ; Li, X. ; Jiao, N. Org. Lett. 2016, 18, 5852.
(q) Xie, S. ; Zhang, Y. ; Ramstrom, O. ; Yan, M. Chem. Sci. 2016, 7, 713.
(r) Yang, S. ; Li, P. ; Wang, Z. ; Wang, L. Org. Lett. 2017, 19, 3386.
(s) Ji, W. ; Li, P. ; Yang, S. ; Wang, L. Chem. Commun. 2017, 53, 8482.
(t) Liu, Y. ; Zhang, Y. ; Wan, J. -P. J. Org. Chem. 2017, 82, 8950.
(u) Yan, Y. ; Zhang, Z. ; Wan, Y. ; Zhang, G. ; Ma, N. ; Liu, Q. J. Org. Chem. 2017, 82, 7957.
(v) Zhang, W. ; Hu, C. ; Zhou, X. Chin. J. Org. Chem. 2017, 37, 1246 (in Chinese).
(张伟, 胡晨旭, 周向葛, 有机化学, 2017, 37, 1246. )
(w) Li, X. ; Jia, X. ; Ma, J. ; Xu, Y. ; Huang, Y. ; Xu, J. Chin. J. Chem. 2017, 35, 984.
(x) Gao, P. ; Bai, Z. Chin. J. Chem. 2017, 35, 1673.
(y) Zhang, J. -R. ; Liao, Y. -Y. ; Deng, J. -C. ; Tang, Z. -L. ; Xu, Y. -L. ; Xu, L. ; Tang, R. -Y. Asian J. Org. Chem. 2017, 6, 305.
(z) Meng, T. ; Feng, C. ; Lantao, L. ; Wang, T. ; Xu, K. ; Zhao, W. Chin. J. Org. Chem. 2016, 36, 1382 (in Chinese).
(孟团结, 冯翠兰, 刘澜涛, 王涛, 许凯, 赵文献, 有机化学, 2016, 36, 1382. )
Valeur, E.; Bradley, M. Chem. Soc. Rev. 2009, 38, 606. doi: 10.1039/B701677H
Pattabiraman, V. R.; Bode, J. W. Nature 2011, 480, 471. doi: 10.1038/nature10702
Schäfer, G.; Matthey, C.; Bode, J. W. Angew. Chem., Int. Ed. 2012, 51, 9173. doi: 10.1002/anie.201204481
Lew, T. T. S.; Lim, D. S. W.; Zhang, Y. Green Chem. 2015, 17, 5140. doi: 10.1039/C5GC01374G
Zhu, Y.-P.; Sergeyev, S.; Franck, P.; Orru, R. V. A.; Maes, B. U. W. Org. Lett. 2016, 18, 4602. doi: 10.1021/acs.orglett.6b02247
Mampuys, P.; Zhu, Y.; Vlaar, T.; Ruijter, E.; Orru, R. V. A.; Maes, B. U. W. Angew. Chem., Int. Ed. 2014, 53, 12849. doi: 10.1002/anie.201406717
Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. J. L.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411. doi: 10.1039/B703488C
Chen, W.; Shao, J.; Hu, M.; Yu, W.; Giulianotti, M. A.; Houghten, R. A.; Yu, Y. Chem. Sci. 2013, 4, 970. doi: 10.1039/C2SC21317F
(a) He, W. ; Li, C. ; Zhang, L. J. Am. Chem. Soc. 2011, 133, 8482.
(b) He, W. ; Xie, L. ; Xu, Y. ; Xiang, J. ; Zhang, L. Org. Biomol. Chem. 2012, 10, 3168.
(c) Xie, L. ; Wu, Y. ; Yi, W. ; Zhu, L. ; Xiang, J. ; He, W. J. Org. Chem. 2013, 78, 9190.
(d) Wu, C. ; Liang, Z. -W. ; Xu, Y. -Y. ; He, W. -M. ; Xiang, J. -N. Chin. Chem. Lett. 2013, 24, 1064.
(e) Li, L. ; Xie, L. ; Wang, F. ; He, W. ; Xiang, J. Chin. J. Org. Chem. 2014, 34, 1864 (in Chinese).
(黎龙香, 谢龙勇, 王风君, 何卫民, 向建南, 有机化学, 2014, 34, 1864. )
(f) Xie, L. ; Yuan, R. ; Wang, R. ; Peng, Z. ; Xiang, J. ; He, W. Eur. J. Org. Chem. 2014, 2668.
(g) Xiang, J. ; Yuan, R. ; Wang, R. ; Yi, N. ; Lu, L. ; Zou, H. ; He, W. J. Org. Chem. 2014, 79, 11378.
(h) Yi, N. ; Wang, R. ; Zou, H. ; He, W. ; Fu, W. ; He, W. J. Org. Chem. 2015, 80, 5023.
(i) Xiang, J. ; Yi, N. ; Wang, R. ; Lu, L. ; Zou, H. ; Pan, Y. ; He, W. Tetrahedron 2015, 71, 694.
(j) Wu, C. ; Yang, P. ; Fu, Z. ; Peng, Y. ; Wang, X. ; Zhang, Z. ; Liu, F. ; Li, W. ; Li, Z. ; He, W. J. Org. Chem. 2016, 81, 10664.
(k) Jiang, J. ; Zou, H. ; Dong, Q. ; Wang, R. ; Lu, L. ; Zhu, Y. ; He, W. J. Org. Chem. 2016, 81, 51.
(l) Zou, H. ; He, W. ; Dong, Q. ; Wang, R. ; Yi, N. ; Jiang, J. ; Pen, D. ; He, W. Eur. J. Org. Chem. 2016, 2016, 116.
(m) Pan, Y. ; Chen, G. -W. ; Shen, C. -H. ; He, W. ; Ye, L. -W. Org. Chem. Front. 2016, 3, 491.
(n) Li, W. ; Yin, G. ; Huang, L. ; Xiao, Y. ; Fu, Z. ; Xin, X. ; Liu, F. ; Li, Z. ; He, W. Green Chem. 2016, 18, 4879.
(o) Xie, L. -Y. ; Duan, Y. ; Lu, L. -H. ; Li, Y. -J. ; Peng, S. ; Wu, C. ; Liu, K. -J. ; Wang, Z. ; He, W. -M. ACS Sustainable Chem. Eng. 2017, 5, 10407.
(p) Wu, C. ; Xin, X. ; Fu, Z. -M. ; Xie, L. -Y. ; Liu, K. -J. ; Wang, Z. ; Li, W. ; Yuan, Z. -H. ; He, W. -M. Green Chem. 2017, 19, 1983.
(q) Xie, L. -Y. ; Li, Y. -J. ; Qu, J. ; Duan, Y. ; Hu, J. ; Liu, K. -J. ; Cao, Z. ; He, W. -M. Green Chem. 2017, 19, 5642.
(r) Xie, L. -Y. ; Qu, J. ; Peng, S. ; Liu, K. -J. ; Wang, Z. ; Ding, M. -H. ; Wang, Y. ; Cao, Z. ; He, W. -M. Green Chem. 2018, 20, 760.
(s) Liu, K. -J. ; Fu, Y. -L. ; Xie, L. -Y. ; Wu, C. ; He, W. -B. ; Peng, S. ; Wang, Z. ; Bao, W. -H. ; Cao, Z. ; Xu, X. ; He, W. -M. ACS Sustainable Chem. Eng. 2018, 6, 4916.
(t) Liu, K. -J. ; Si, J. ; Lu, L. -H. ; Tang, L. -L. ; Tang, S. -S. ; Tang, H. -S. ; Tang, Z. ; He, W. -M. ; Xu. X. Green Chem. 2018, 10. 1039/C8GC00223A.
Han, K.-J.; Tae, B. S.; Kim, M. Org. Prep. Proced. Int. 2005, 37, 198. doi: 10.1080/00304940509354888
Tsukamoto, H.; Suzuki, R.; Kondo, Y. J. Comb. Chem. 2006, 8, 289. doi: 10.1021/cc0600066
Ramalingan, C.; Park, Y.-T. J. Org. Chem. 2007, 72, 4536. doi: 10.1021/jo070297k
Hosseini-Sarvari, M.; Sodagar, E.; Doroodmand, M. M. J. Org. Chem. 2011, 76, 2853. doi: 10.1021/jo2002769
Chiang, P.-C.; Kim, Y.; Bode, J. W. Chem. Commun. 2009, 4566.
Jeddeloh, M. R.; Holden, J. B.; Nouri, D. H.; Kurth, M. J. J. Comb. Chem. 2007, 9, 1041. doi: 10.1021/cc700117a
Furuya, Y.; Ishihara, K.; Yamamoto, H. J. Am. Chem. Soc. 2005, 127, 11240. doi: 10.1021/ja053441x
Youn, S. W.; Bihn, J. H.; Kim, B. S. Org. Lett. 2011, 13, 3738. doi: 10.1021/ol201416u
Yang, Y.-H.; Shi, M. J. Org. Chem. 2005, 70, 8645. doi: 10.1021/jo0516988
Ye, W.; Mo, J.; Zhao, T.; Xu, B. Chem. Commun. 2009, 3246.
Wu, W.; Zhang, Z.; Liebeskind, L. S. J. Am. Chem. Soc. 2011, 133, 14256. doi: 10.1021/ja2065158
Zhang, L.; Su, S.; Wu, H.; Wang, S. Tetrahedron 2009, 65, 10022. doi: 10.1016/j.tet.2009.09.101
Wang, Y.; Zhu, D.; Tang, L.; Wang, S.; Wang, Z. Angew. Chem., Int. Ed. 2011, 50, 8917. doi: 10.1002/anie.v50.38
Lagerlund, O.; Larhed, M. J. Comb. Chem. 2006, 8, 4. doi: 10.1021/cc050102r
Jørgensen, C. G.; Frølund, B.; Kehler, J.; Jensen, A. A. ChemMedChem 2011, 6, 725. doi: 10.1002/cmdc.v6.4
Kumar, S.; Singh, H. B. J. Chem. Sci. 2005, 117, 621. doi: 10.1007/BF02708290
Antonchick, A. P.; Samanta, R.; Kulikov, K.; Lategahn, J. Angew. Chem., Int. Ed. 2011, 50, 8605. doi: 10.1002/anie.201102984
Liu, H.; Wang, X.; Gu, Y. Org. Biomol. Chem. 2011, 9, 1614. doi: 10.1039/c0ob00749h
Prakash, G. K. S.; Moran, M. D.; Mathew, T.; Olah, G. A. J. Fluorine Chem. 2009, 130, 806. doi: 10.1016/j.jfluchem.2009.05.015
O'Conner, C.; McLennan, D.; Calvert, D.; Lomax, T.; Porter, A.; Rogers, D. Aust. J. Chem. 1984, 37, 497. doi: 10.1071/CH9840497
Chrétien, J.-M.; Zammattio, F.; Le Grognec, E.; Paris, M.; Cahingt, B.; Montavon, G.; Quintard, J.-P. J. Org. Chem. 2005, 70, 2870. doi: 10.1021/jo0480141
Knauber, T.; Arikan, F.; Röschenthaler, G.-V.; Gooßen, L. J. Chem.-Eur. J. 2011, 17, 2689. doi: 10.1002/chem.201002749
Morimoto, H.; Tsubogo, T.; Litvinas, N. D.; Hartwig, J. F. Angew. Chem., Int. Ed. 2011, 50, 3793. doi: 10.1002/anie.v50.16
Shuai, Q.; Deng, G.; Chua, Z.; Bohle, D. S.; Li, C.-J. Adv. Synth. Catal. 2010, 352, 632. doi: 10.1002/adsc.v352:4
Kim, B. S.; Jang, C.; Lee, D. J.; Youn, S. W. Chem. Asian J. 2010, 5, 2336. doi: 10.1002/asia.v5:11
Heys, J. R.; Elmore, C. S. J. Labelled Compd. Radiopharm. 2009, 52, 189. doi: 10.1002/jlcr.v52:6
Hosseinzadeh, R.; Tajbakhsh, M.; Mohadjerani, M.; Ghorbani, E. Chin. J. Chem. 2008, 26, 2120. doi: 10.1002/cjoc.v26:11
Table 1. Screening the optimized reaction conditionsa
|
|||
| Entry | Catalyst (equiv.) | Solvent | Yieldb/% |
| 1 | Et3N (1.5) | DMF | 11 |
| 2 | DBU (1.5) | DMF | 68 |
| 3 | DBN (1.5) | DMF | 90 |
| 4 | i-Pr2NEt (1.5) | DMF | Trace |
| 5 | Na2CO3 (1.5) | DMF | 6 |
| 6 | Cs2CO3 (1.5) | DMF | 9 |
| 7 | KF-Celite (1.5) | DMF | Trace |
| 8c | Amberlite IRA-4200 (10 wt%) | DMF | N.R. |
| 9 | DBN (2) | DMF | 97 |
| 10 | DBN (1.2) | DMF | 78 |
| 11 | DBN (0.2) | DMF | 12 |
| 12 | — | DMF | N.R. |
| 13 | DBN (2) | DCM | 8 |
| 14 | DBN (2) | THF | 15 |
| 15 | DBN (2) | MeCN | 72 |
| 16 | DBN (2) | Toluene | Trace |
| 17 | DBN (2) | DMSO | Trace |
| 18 | DBN (2) | MeOH | Trace |
| 19d | DBN (2) | DMF | 91 |
| 20e | DBN (2) | DMF | 94 |
| a Unless otherwise specified, the reactions were carried out in a vial in the presence of 1a (0.15 mmol), 2a (0.1 mmol), base, solvent (1 mL). b Estimated by 1H NMR spectroscopy using diethyl phthalate as an internal reference. c The quantity of Amberlite IRA-4200 was 0.1 g. d The reaction was conducted under 40 kHz/30 W ultrasonic radiation for 1 h. e The reaction was conducted under 150 W microwave radiation of 20 min. N.R.: no reaction. | |||
 下载: 导出CSV
下载: 导出CSV
Table 2. Reaction scope of carboxylic acidsa
|
|
| a All reactions were carried out in a vail in the presence of 1 (0.45 mmol), 2 (0.3 mmol), DBN (0.6 mmol) and DMF (3 mL); isolated yields are reported. |
下载: 导出CSV
Table 3. Reaction scope of isothiocyanatesa
|
|
| a All reactions were carried out in a vail in the presence of 1 (0.45 mmol), 2 (0.3 mmol), DBN (0.6 mmol) and DMF (3 mL); isolated yields are reported. |
下载: 导出CSV
Table 4. Reaction scope of late-stage modificationa
|
| a All reactions were carried out in a vail in the presence of 1 (0.45 mmol), 2 (0.3 mmol), DBN (0.6 mmol) and DMF (3 mL); isolated yields are reported. |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们