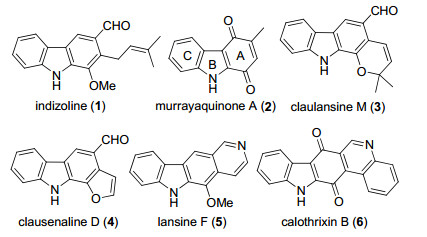
图 1.
代表性咔唑生物碱
Figure 1.
Representative carbazole alkaloids

咔唑类天然产物是一类重要的含氮杂环化合物, 广泛存在于九里香属、山小橘属、黄皮属等芸香科植物中[1].此外, 在一些菌类[2, 3]和藻类[4]中也发现了咔唑类生物碱.咔唑类生物碱具有抗菌、抗炎、抗氧化、抗癌和抗老年痴呆等多种生物活性[1], 并且在有机光电材料和配位化学等领域也有着广泛的应用[5, 6].咔唑类生物碱结构多种多样, 除基本的三环结构外, 还包括吡喃并咔唑生物碱、呋喃并咔唑生物碱、吡啶并咔唑生物碱、喹啉并咔唑生物碱等多种类型(图 1).其中, 三环咔唑醌类生物碱Murrayaquinone A是从九里香属植物中分离得到[7], 具有强心活性[8].虽然其结构相对简单, 但在合成上却受到很大的重视, 几乎所有咔唑化合物合成新方法在该类天然产物全合成中的应用都以其作为首选目标分子.目前已有多个课题组报道了Murrayaquinone A的全合成, 根据合成过程中最后一个环的构建, 可以分为吡咯B环的构建, 以及苯醌A环的构建两种策略.其中, 绝大多数报道都是采用构建吡咯B环的策略, 包括过渡金属催化的二苯胺衍生物的偶联[9]、2-氨基醌和苯炔或环己烯酮的成环反应[10]以及2-硝基苯乙烯衍生物的还原环化[11]等; 而以包含B、C环的吲哚衍生物为底物来构建A环的报道较少, 主要有烯烃复分解[12]、[4+2]环加成[13]等方法.此外, 也有对已有咔唑环进行修饰从而合成Murrayaquinone A的报道[14].
我们课题组长期从事具有生物活性的天然产物的全合成[15].在咔唑类天然产物全合成方面, 我们发展了三氟乙酸酐介导的直接以羧酸作为酰化剂[16]的分子内酰化策略构建了咔唑的A环, 进而完成了多个咔唑类天然产物的全合成[15i~15k].我们现在报道一种新的通过钯催化的分子内吲哚α-C—H烯基化构建咔唑A环, 从而实现Murrayaquinone A的全合成的策略.
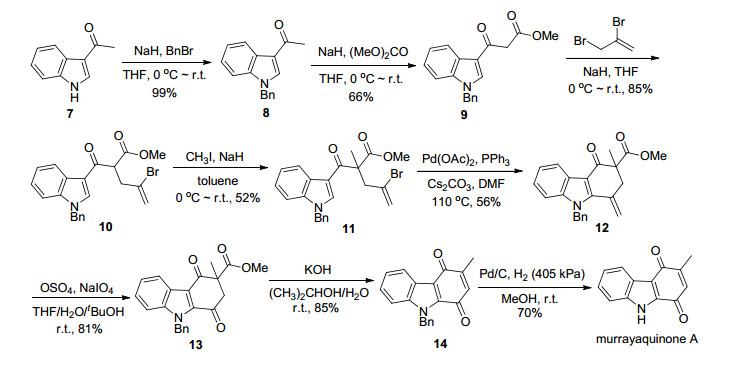
如Scheme 1所示, 以3-乙酰基吲哚(7)为起始原料, 吲哚氮原子用苄基保护得化合物8.化合物8在氢化钠的作用下与碳酸二甲酯反应, 在乙酰基的α-碳上引入酯基, 以66%的产率得到β-二羰基化合物9.化合物9先后与2, 3-二溴丙烯、碘甲烷发生亲核取代反应得到化合物11.化合物11在醋酸钯、三苯基膦和碳酸铯的存在下发生分子内吲哚α-C—H烯基化[17, 18]反应, 以56%的收率得到四氢咔唑酮12.需要指出的是, 该反应过程中并没有双键发生异构化的产物生成.化合物12的碳-碳双键经四氧化锇和高碘酸钠氧化以81%的收率得到二氢咔唑醌13.化合物13经氢氧化钾处理脱掉酯基, 并在反应条件下原位氧化生成咔唑醌14.最后经催化氢化脱掉苄基保护基得目标产物Murrayaquinone A.
以3-乙酰基吲哚为原料, 以钯催化的分子内吲哚α-C—H烯基化作为关键步骤来构建咔唑A环, 经8步反应合成得到了咔唑天然产物Murrayaquinone A.这种构建咔唑A环的方法是首次被用于该类天然产物的全合成, 且关环产物12的C-1、C-2、C-4位非常容易进一步官能团化, 从而为其他相关化合物的合成提供了一种新的思路.
核磁共振波谱仪: Bruker AV 400 MHz (内标: TMS); 熔点仪: XT4A显微熔点测定仪(北京科仪电光仪器厂); 四极杆质谱仪: Micromass Q-TOF; 薄层色谱层析硅胶: GF254(青岛海洋化工厂分厂); 柱层析硅胶: 200~300目(山东省烟台市牟平区康必诺化学试剂厂).所用试剂均为分析纯试剂.
称取3-乙酰基吲哚(10.0 g, 62.8 mmol)于250 mL圆底烧瓶中, 加入100 mL无水四氢呋喃, 搅拌溶解.将体系冷却至0 ℃, 然后分5次缓慢加入氢化钠(60% dispersion in mineral oil, 5.0 g, 125.0 mmol).加毕后, 继续搅拌30 min.缓慢滴加溴化苄(8.0 mL, 67.0 mmol).滴加完后, 反应体系缓慢升至室温, 搅拌12 h, 薄层色谱(TLC)监测反应完全.小心加入冰水淬灭反应, 旋蒸除去四氢呋喃, 乙酸乙酯萃取(20 mL×3).合并有机相, 无水硫酸钠干燥, 过滤, 旋干.硅胶柱层析分离[V(乙酸乙酯):V(石油醚)=1:4]得白色固体8 (15.5 g, 62.2 mmol), 产率99%. m.p. 111~113 ℃ (lit.[19] m.p. 109~110 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.41~8.39 (m, 1H), 7.73 (s, 1H), 7.35~7.22 (m, 6H), 7.15~7.12 (m, 2H), 5.31 (s, 2H), 2.49 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 193.0, 137.0, 135.8, 135.0, 129.0, 128.2, 126.9, 126.4, 123.4, 122.6, 122.6, 117.4, 110.1, 50.7, 27.6; HRMS (ESI) calcd for C17H15NNaO [M+Na]+ 272.1046, found 272.1048.
称取化合物8 (2.9 g, 11.6 mmol)于100 mL圆底烧瓶中, 加入30 mL无水四氢呋喃, 搅拌溶解.将体系冷却至0 ℃, 然后分3次缓慢加入氢化钠(60% dispersion in mineral oil, 1.4 g, 35.4 mmol).加毕后, 继续搅拌30 min.缓慢滴加碳酸二甲酯(2.0 mL, 23.6 mmol), 滴加完后, 反应体系缓慢升至室温, 搅拌8 h, TLC监测反应完全.冰水淬灭, 旋蒸除去四氢呋喃, 乙酸乙酯萃取(5 mL×3).合并有机相, 无水硫酸钠干燥, 过滤, 旋干.硅胶柱层析分离[V(乙酸乙酯):V(石油醚)=1:2]得白色固体9 (2.4 g, 7.8 mmol), 产率66%. m.p. 129~130 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.40~8.37 (m, 1H), 7.81 (s, 1H), 7.36~7.25 (m, 6H), 7.16~7.14 (m, 2H), 5.33 (s, 2H), 3.86 (s, 2H), 3.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ:186.3, 168.5, 137.1, 135.8, 135.5, 129.1, 128.3, 127.0, 126.5, 123.9, 123.1, 122.7, 116.4, 110.3, 52.4, 50.9, 47.2; IR (neat) νmax: 1736, 1639, 1390 cm-1; HRMS (ESI) calcd for C19H17NNaO3 [M+Na]+ 330.1101, found 330.1107.
称取化合物9 (1.7 g, 5.5 mmol)于100 mL圆底烧瓶中, 加入50 mL无水四氢呋喃, 搅拌溶解.将体系冷却至0 ℃, 然后分3次缓慢加入氢化钠(60% dispersion in mineral oil, 0.23 g, 5.7 mmol).加毕后, 继续搅拌30 min.缓慢滴加2, 3-二溴-1-丙烯(0.72 mL, 6.3 mmol), 滴加完后, 反应体系缓慢升至室温, 搅拌12 h, TLC监测反应完全.冰水淬灭, 旋蒸除去四氢呋喃, 乙酸乙酯萃取(10 mL×3).合并有机相, 无水硫酸钠干燥, 过滤, 旋干.硅胶柱层析分离[V(乙酸乙酯):V(石油醚)=1:6]得无色油状物10 (2.0 g, 4.7 mmol), 产率85%. 1H NMR (400 MHz, CDCl3) δ: 8.43~8.41 (m, 1H), 8.00 (s, 1H), 7.36~7.23 (m, 6H), 7.18~7.16 (m, 2H), 5.69 (d, J=1.6 Hz, 1H), 5.38 (d, J=1.6 Hz, 1H), 5.34 (s, 2H), 4.52 (t, J=7.1 Hz, 1H), 3.65 (s, 3H), 3.18 (dd, J=14.7, 7.2 Hz, 1H), 3.13 (dd, J=14.7, 7.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 187.3, 169.5, 137.1, 136.2, 135.3, 130.2, 129.0, 128.3, 127.1, 126.6, 123.9, 123.2, 122.8, 119.9, 116.1, 110.3, 53.7, 52.5, 50.9, 40.7; IR (neat) νmax: 1739, 1645, 1526, 1386, 1186 cm-1; HRMS (ESI) calcd for C22H20NNaO3 [M+Na]+ 448.0519, found 448.0511.
称取化合物10 (792 mg, 1.9 mmol)于25 mL圆底烧瓶中, 加入10 mL甲苯溶解.将体系冷却至0 ℃, 然后分3次缓慢加入氢化钠(60% dispersion in mineral oil, 298 mg, 7.4 mmol).加毕后, 继续搅拌30 min.缓慢滴加碘甲烷(0.23 mL, 3.7 mmol).滴加完后, 反应体系缓慢升至室温, 搅拌15 h后, TLC监测反应完全.冰水淬灭, 旋蒸除去甲苯, 乙酸乙酯萃取(5 mL×3).合并有机相, 无水硫酸钠干燥, 过滤, 旋干.硅胶柱层析分离[V(乙酸乙酯):V(石油醚)=1:4]得无色粘稠油状物11 (426 mg, 0.97 mmol), 产率52%. 1H NMR (400 MHz, CDCl3) δ: 8.49~8.47 (m, 1H), 7.82 (s, 1H), 7.36~7.28 (m, 6H), 7.13~7.11 (m, 2H), 5.66~5.65 (m, 1H), 5.59 (d, J=1.6 Hz, 1H), 5.36 (d, J=15.8 Hz, 1H), 5.31 (d, J=15.8 Hz, 1H), 3.59 (s, 3H), 3.37 (d, J=15.0 Hz, 1H), 3.28 (d, J=15.0 Hz, 1H), 1.69 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 190.8, 174.7, 136.3, 135.7, 133.7, 129.1, 128.3, 128.2, 127.9, 126.9, 123.8, 123.2, 123.1, 121.8, 113.4, 110.1, 57.2, 52.5, 50.8, 47.1, 21.4; IR (neat) νmax: 1731, 1645, 1526, 1454, 1381 cm-1; HRMS (ESI) calcd for C23H22BrNNaO3 [M+Na]+ 462.0675, found 462.0685.
称取化合物11 (380 mg, 0.86 mmol)于35 mL的密封管中, 加入15 mL无水N, N-二甲基甲酰胺溶解, 然后加入碳酸铯(560 mg, 1.72 mmol), 三苯基膦(23 mg, 0.09 mmol).加毕, 用油泵抽真空, 换氮气3次.再加入醋酸钯(20 mg, 0.09 mmol), 重复抽真空, 换氮气3次, 最后拧紧密封管旋塞, 置于110 ℃油浴中反应. 21 h后, TLC监测反应完全, 加入饱和食盐水稀释, 乙酸乙酯萃取(10 mL×3).合并有机相, 用饱和食盐水洗3次, 无水硫酸钠干燥, 过滤, 旋干.硅胶柱层析分离[V(乙酸乙酯):V(石油醚)=1:6]得黄色粘稠状物12 (174 mg, 0.48 mmol), 产率56%. 1H NMR (400 MHz, CDCl3) δ: 8.39~8.37 (m, 1H), 7.36~7.27 (m, 5H), 7.23~7.21 (m, 1H), 7.09~7.07 (m, 2H), 5.55 (s, 2H), 5.49 (s, 1H), 5.35 (s, 1H), 3.72 (s, 3H), 3.43 (d, J=13.4 Hz, 1H), 2.76 (d, J=13.4 Hz, 1H), 1.56 (s, 3H); 13C NMR (100 MHz, CDCl3)δ: 191.9, 173.3, 145.7, 139.3, 136.1, 132.2, 129.2, 127.8, 125.7, 124.8, 124.8, 123.5, 122.5, 116.6, 112.4, 110.3, 56.0, 52.5, 48.5, 46.1, 20.4; IR (neat) νmax: 1736, 1646, 1453, 1244 cm-1; HRMS (ESI) calcd for C23H21N- NaO3 [M+Na]+ 382.1414, found 382.1406.
称取化合物12 (51 mg, 0.14 mmol)溶于四氢呋喃(6 mL)与水(2 mL)的混合溶剂中, 然后依次加入高碘酸钠(180 mg, 0.84 mmol)和四氧化锇的叔丁醇溶液(9 mg, 0.3 mL, 0.08 mmol).室温搅拌过夜, 反应完全后加入饱和连二亚硫酸钠溶液, 旋蒸除去叔丁醇和四氢呋喃, 乙酸乙酯萃取(5 mL×3).合并有机相, 用饱和食盐水洗3次, 无水硫酸钠干燥, 过滤, 旋干.硅胶柱层析分离[V(乙酸乙酯):V(石油醚)=1:2]得无色油状物13 (41 mg, 0.11 mmol), 产率81%. 1H NMR (400 MHz, CDCl3) δ: 8.38~8.36 (m, 1H), 7.45~7.43 (m, 2H), 7.41~7.36 (m, 1H), 7.30~7.23 (m, 3H), 7.15~7.13 (m, 2H), 5.90 (s, 2H), 3.66 (s, 3H), 3.43 (d, J=16.3 Hz, 1H), 2.98 (d, J=16.3 Hz, 1H), 1.66 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 190.3, 189.1, 171.9, 139.4, 136.4, 136.2, 128.8, 127.8, 127.7, 126.8, 124.5, 123.7, 123.6, 119.7, 111.5, 58.8, 53.3, 50.1, 48.2, 20.9; IR (neat) νmax: 1737, 1672, 1196 cm-1; HRMS (ESI) calcd for C22H19NNaO4 [M+Na]+ 384.1206, found 384.1202.
称取化合物13 (45 mg, 0.12 mmol)溶于异丙醇(4 mL)与水(1 mL)的混合溶剂中, 然后加入氢氧化钾(168 mg, 3.0 mmol).室温搅拌4 h后, 用2 mol/L盐酸淬灭, 乙酸乙酯萃取(5 mL×3).合并有机相, 用饱和食盐水洗涤3次, 无水硫酸钠干燥, 过滤, 旋干.硅胶柱层析分离[V(乙酸乙酯):V(石油醚)=1:4]得黄色固体14 (32 mg, 0.11 mmol), 收率85%. m.p. 110~112 ℃ (lit.[14b] m.p. 157 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.33~8.31 (m, 1H), 7.44~7.34 (m, 3H), 7.30~7.27 (m, 1H), 7.25~7.24 (m, 2H), 7.17~7.15 (m, 2H), 6.45 (q, J=1.6 Hz, 1H), 5.86 (s, 2H), 2.15 (d, J=1.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 183.7, 181.2, 147.5, 139.0, 136.4, 133.5, 132.9, 128.8, 127.8, 126.9, 126.8, 124.6, 123.9, 123.2, 117.3, 111.5, 48.1, 15.8; HRMS (ESI) calcd for C20H15NNaO2 [M+Na]+ 324.0995, found 324.0991.
称取化合物14 (30 mg, 0.1 mmol)于50 mL高压釜中, 加入10 mL甲醇溶解, 然后加入10%钯/碳(11 mg, 0.01 mmol).先用氮气换气三次, 再用氢气换气三次.氢气压力405 kPa下, 室温反应5 h.过滤, 滤液旋干.硅胶柱层析分离[V(乙酸乙酯):V(石油醚)=1:4]得红色固体产品(15 mg, 70%), 收率70%. m.p. 242~243 ℃ (lit.[9a] m.p. 237~239 ℃); 1H NMR (400 MHz, DMSO- d6) δ: 12.82 (s, 1H), 8.05 (d, J=8.0 Hz, 1H), 7.54 (d, J=8.2 Hz, 1H), 7.39 (td, J=7.2, 1.2 Hz, 1H), 7.34~7.30 (m, 1H), 6.62 (q, J=1.6 Hz, 1H), 2.07 (d, J=1.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 183.5, 180.5, 148.4, 137.9, 136.3, 132.1, 126.7, 124.3, 124.0, 122.1, 115.9, 114.3, 16.1; HRMS (ESI) calcd for C17H15N2 [M+H]+ 212.0712, found 212.0718.
辅助材料(Supporting Information)化合物8~14和2的核磁共振谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Knölker, H.-J.; Reddy, K. R. Chem. Rev. 2002, 102, 4303.
(b) Schmidt, A. W.; Reddy, K. R.; Knölker, H.-J. Chem. Rev. 2012, 112, 3193.
Ruanpanun P.; Dame, Z. T.; Laatsch, H.; Lumyong, S. FEMS Microbiol. Lett. 2011, 322, 77. doi: 10.1111/fml.2011.322.issue-1
Hoshino, S.; Zhang, L.; Awakawa, T.; Wakimoto, T.; Onaka, H.; Abe, I. J. Antibiot. 2015, 68, 342. doi: 10.1038/ja.2014.147
Liu, D. Q.; Mao, S. C.; Zhang, H. Y.; Yu, X. Q.; Feng, M. T.; Wang, B.; Feng, L. H.; Guo, Y. W. Fitoterapia 2013, 91, 15. doi: 10.1016/j.fitote.2013.08.014
张飞飞, 周成合, 颜建平, 有机化学, 2010, 30, 783. http://sioc-journal.cn/Jwk_yjhx/CN/Y2010/V30/I06/783Zhang, F. F.; Zhou, C. H.; Yan, J. P. Chin. J. Org. Chem. 2010, 30, 783(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2010/V30/I06/783
刘勇, 张峰, 邹林波, 蹇军友, 鲍小平, 有机化学, 2013, 33, 2485. http://sioc-journal.cn/Jwk_yjhx/CN/Y2013/V33/I12/2485Liu, Y.; Zhang, F.; Zou, L. B.; Jian, J. Y.; Bao, X. P. Chin. J. Org. Chem. 2013, 33, 2485(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2013/V33/I12/2485
Wu, T. S.; Ohta, T.; Furukawa, H.; Kuoh, C. S. Heterocycles 1983, 20, 1267. doi: 10.3987/R-1983-07-1267
Takeya, K.; Itoigawa, M.; Furukawa, H. Eur. J. Pharmacol. 1989, 169, 137. doi: 10.1016/0014-2999(89)90825-X
(a) Yogo, M.; Ito, C.; Furukawa, H. Chem. Pharm. Bull. 1991, 39, 328.
(b) Hagelin, H.; Oslob, J. D.; Å kermark, B. Chem. Eur. J. 1999, 5, 2413.
(c) Norcott, P.; Spielman, C.; McErlean, C. S. P. Green Chem. 2012, 14, 605.
(d) Bedford, R. B.; Bowen, J. G.; Weeks, A. L. Tetrahedron 2013, 69, 4389.
(e) Kaliyaperumal, S. A.; Banerjee, S.; Kumar, U. K. S. Org. Biomol. Chem. 2014, 12, 6105.
(a) Guo, J.; Kiran, I. N. C.; Reddy, R. S.; Gao, J.; Tang, M.; Liu, Y.; He, Y. Org. Lett. 2016, 18, 2499.
(b) McDonald, J. W.; Miller, J. E.; Kim, M.; Velu, S. E. Tetrahedron Lett. 2018, 59, 550.
Scott, T. L.; Söderberg, B. C. G. Tetrahedron 2003, 59, 6323. doi: 10.1016/S0040-4020(03)00976-1
(a) Nishiyama, T.; Choshi, T.; Kitano, K.; Hibino, S. Tetrahedron Lett. 2011, 52, 3876.
(b) Nishiyama, T.; Hatae, N.; Yoshimura, T.; Takaki, S.; Abe, T.; Ishikura, M.; Hibino, S.; Choshi, T. Eur. J. Med. Chem. 2016, 121, 561.
Mal, D.; Senapati, B. K.; Pahari, P. Tetrahedron 2007, 63, 3768. doi: 10.1016/j.tet.2007.02.060
(a) Matsuo, K.; Ishida, S. Chem. Pharm. Bull. 1994, 42, 1325.
(b) Bhosale, S. M.; Momin, A. A.; Kusurkar, R. S. Tetrahedron 2012, 68, 6420.
(a) Hong, M.; Liu, H.; Sun, L.; Jia, F.; Liu, Y.; Jiang, Q.; Song, C.; Chang, J. Synlett 2011, 2995.
(b) Song, C.; Liu, H.; Hong, M.; Liu, Y.; Jia, F.; Sun, L.; Pan, Z.; Chang, J. J. Org. Chem. 2012, 77, 704.
(c) Chang, J.; Wang, S.; Shen, Z.; Huang, G.; Zhang, Y.; Zhao, J.; Li, C.; Fan, F.; Song, C. Tetrahedron Lett. 2012, 53, 6755.
(d) Fan, F.; Dong, J.; Wang, J.; Song, L.; Song, C.; Chang, J. Adv. Synth. Catal. 2014, 356, 1337.
(e) Wang, J.; Wang, J.; Li, C.; Meng, Y.; Wu, J.; Song, C.; Chang, J. J. Org. Chem. 2014, 79, 6354.
(f) Li, E.; Li, C.; Wang, J.; Wang, J.; Dong, L.; Guo, X.; Song, C.; Chang, J. Tetrahedron 2014, 70, 874.
(g) Meng, Y.; Liu, Y.; Lv, Z.; Wang, J.; Wang, Y.; Song, C.; Chang, J. Nat. Prod. Commun. 2015, 10, 2031.
(h) Ding, Q.; Zhang, Y.; Shen Z.; Song, C.; Chang, J. Helv. Chim. Acta 2015, 98, 128.
(i) Liu, Y.; Guo, Y.; Ji, F.; Gao, D.; Song, C.; Chang, J. J. Org. Chem. 2016, 81, 4310.
(j) Guo, Y.; Huang, H.; Zhang, T.; Qi, S.; Liu, Y.; Li, M.; Wang, P.; Song, C.; Chang, J. Asian J. Org. Chem. 2016, 5, 1269.
(k) Ji, F.; Huang, H.; Li, M.; Song, C.; Chang, J. Synthesis 2018, 50, 3921.
(a) Song, C.; Knight, D. W.; Whatton, M. A. Tetrahedron Lett. 2004, 45, 9573.
(b) Song, C.; Zhao, P.; Liu, Y.; Liu, H.; Li, W.; Shi, S.; Chang, J. Tetrahedron 2010, 66, 5378.
(c) Song, C.; Liu, Y.; Zhao, P.; Sun, X.; Li, W.; Liu, H.; Chang, J. Synthesis 2011, 45.
(d) Chang, J.; Sun, L.; Dong, J.; Shen, Z.; Zhang, Y.; Wu, J.; Wang, R.; Wang, J.; Song, C. Synlett 2012, 2704.
(e) Wang, S.; Yang, Q.; Dong, J.; Li, C.; Sun, L.; Song, C.; Chang, J. Eur. J. Org. Chem. 2013, 7631.
(a) Ferreira, E. M.; Stoltz, B. M. J. Am. Chem. Soc. 2003, 125, 9578.
(b) Liu, C.; Han, X.; Wang, X.; Widenhoefer, R. A. J. Am. Chem. Soc. 2004, 126, 3700.
(c) Liu, C.; Widenhoefer, R. A. J. Am. Chem. Soc. 2004, 126, 10250.
(d) Han, X.; Widenhoefer, R. A. Org. Lett. 2006, 8, 3801.
(e) Han, X.; Lu, X. Org. Lett. 2009, 11, 2381.
(f) Bandini, M.; Bottoni, A.; Chiarucci, M.; Cera, G.; Miscione, G. P. J. Am. Chem. Soc. 2012, 134, 20690.
(g) Schiffner, J. A.; Wö ste, T. H.; Oestreich, M. Eur. J. Org. Chem. 2010, 174.
(h) Zhang, T.; Xu, H.; Song, C.; Chang, J. Tetrahedron Lett. 2018, 59, 4562.
(a) Chen, P.; Meng, Y.; Wang, H.; Han, F.; Wang, Y.; Song, C.; Chang, J. Org. Lett. 2016, 18, 3914.
(b) Zhang, T.; Song, C.; Meng, Y.; Chen, P.; Xu, H.; Chang, J. J. Org. Chem. 2017, 82, 9905.
Sechi, M.; Derudas, M.; Dallocchio, R.; Dessì, A.; Bacchi, A.; Sannia, L.; Carta, F.; Palomba, M.:Ragab, O.; Chan, C.; Shoemaker, R.; Sei, S.; Dayam, R.; Neamati, N. J. Med. Chem. 2004, 47, 5298. doi: 10.1021/jm049944f

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
