Table 1.
Crystallographic data and refinement parameters for complexes 1~3
Citation:
ZHANG Mei-Li, ZHENG Yan-Jin, LIU Min, REN Yi-Xia, WANG Ji-Jiang, CUI Hua-Li, LIU Lin. Crystal Structure and Electrochemistry Properties of Three Co(Ⅱ) Complexes Based on Flexible Phenylenediacetate Ligands[J]. Chinese Journal of Inorganic Chemistry,
2019, 35(10): 1813-1820.
doi:
10.11862/CJIC.2019.214

基于柔性苯二乙酸构筑的三个钴配合物的晶体结构和电化学性质
English
Crystal Structure and Electrochemistry Properties of Three Co(Ⅱ) Complexes Based on Flexible Phenylenediacetate Ligands
Abstract:
Three new coordination complexes, {[Co1.5(opda)1.5(mbib)2(H2O)2]·H2O}n (1), {[Co(mpda)(mbib)]·H2O}n (2) and {[Co(ppda)(mbib)]·H2O}n (3) (H2opda=1, 2-phenylenediacetic acid, H2mpda=1, 3-phenylenediacetic acid, H2ppda=1, 4-phenylenediacetic acid, mbib=1, 3-bis(1-imidazoly)benzene), have been synthesized by using cobalt salt and bis(imidazole) ligand in the presence of different flexible phenylenediacetate blocks under hydrothermal conditions, and characterized by elemental analysis, IR spectroscopy and single-crystal X-ray crystallography. Single-crystal X-ray diffraction analysis revealed that the diverse structures of these complexes are mainly attributed to various coordination geometries of the central cobalt(Ⅱ) cations, coordination modes and conformations of the flexible phenylenediacetic acid and bis(imidazole) ligands. Furthermore, thermal gravimetric analysis (TGA) and the electrochemistry properties of 1~3 are also investigated, and all of them have good cyclic voltammograms.
-
Key words:
- coordination polymer
- / crystal structure
- / electrochemistry property
-
0. Introduction
The rational design and synthesis of polymeric metal-organic hybrid complexes has been extensively studied in recent years, not only on their intriguing structural diversity[1], but also on their intrinsic aesthetic appeal and encouraging properties such as catalysis[2], luminescence[3-7], chemical sensing[8], energy storage conversion[9], and drug delivery[10-11]. To date, it is still a challenge to predict the exact structure of assembly products in crystal engineering since the structure is mainly dependent by many factors such as the coordination geometries of the metal centers, the coordination behaviours of the organic building blocks, and guest solvent or counter ions[12].
Among the reported studies, organic aromatic polycarboxylate ligands have attracted intensive research interest due to their various coordination modes to metal ions, resulting from completely or partially deprotonated sites allowing for the large diversity of topologies[13]. In contrast to those of rigid ligands, the backbone flexibility and conformation changeability afford these flexible ligands versatile bridging modes and make the structures of their coordination polymers more diversiform and more difficult to predict[14-15]. However, it is still a challenge to understanding the control of structure and topology of coordination polymers. In our recent work, a series of coordination polymers based on flexible phenylene-diacetate and different bis(imidazole) ligands have been successfully obtained[16-17], and we are systemati-cally investigating the influence of the coordination chemistry of phenylenediacetate ligands with different coordination groups, conformations, and flexibility. As a continuation of our research, by employing the mixture of different flexible phenylenediacetate building blocks in the presence of bis(imidazole) ligand 1, 3-bis(1-imidazoly)benzene (mbib), three new coordination polymers {[Co1.5(opda)1.5(mbib)2(H2O)2]·H2O}n (1), {[Co(mpda)(mbib)]·H2O}n (2) and {[Co(ppda)(mbib)]·H2O}n (3) (H2opda=1, 2-phenylenediacetic acid, H2mpda=1, 3-phenylenediacetic acid, H2ppda=1, 4-phenylenediacetic acid, showing structural diversities have been synthesized and structurally characterized. Furthermore, their thermal gravimetric analysis (TGA) and electrochemistry properties have also been investigated.
1. Experimental
1.1 Materials and methods
All the starting reagents and solvents were commercially available and used as received without further purification. The hydrothermal reaction was performed in a 25 mL Teflon-lined stainless steel autoclave under autogenous pressure. Elemental analyses for C, H, and N were carried out on a Flash 2000 elemental analyzer. Thermal gravimetric analysis (TGA) were carried out on a SDT Q600 thermogravi-metric analyzer. A platinum pan was used for heating the sample with a heating rate of 10 ℃·min-1 under a N2 atmosphere. Powder X-ray diffraction (PXRD) measurements were performed on a Bruker D8-ADVANCE X-ray diffractometer with Cu Kα radiation (λ=0.154 2 nm), and the experimental test conditions were the voltage of 40 kV, the current of 30 mA, and the scanning range of 5°~50°.
1.2 Syntheses of the complexes
{[Co1.5(opda)1.5(mbib)2(H2O)2]·H2O}n (1). A mixture of H2opda (0.009 6 g, 0.05 mmol), Co(OAc)2·4H2O (0.024 9 g, 0.1 mmol), (0.018 6 g, 0.1 mmol) and KOH (5.6 mg, 0.1 mmol) were added to water (12 mL) in a 25 mL Teflon-lined stainless steel vessel. The mixture was heated at 150 ℃ for 72 h. After the reactive mixture was slowly cooled to room temperature, violet block crystals of 1 were obtained. Elemental analysis Calcd. for C78H76Co3N16O18(%): C 55.03, H 4.50, N 13.17; Found(%): C 55.37, H 4.25, N 13.53.
{[Co(mpda)(mbib)]·H2O}n (2). Complex 2 was synthesized by a procedure similar to that of 1, except that H2mpda (0.009 6 g, 0.05 mmol) replaced H2opda. Violet block crystals of 2 were obtained. Elemental analysis Calcd. for C44H40Co2N8O10(%): C 55.12, H 4.21, N 11.69; Found(%): C 54.89, H 4.35, N 11.37.
{[Co(ppda)(mbib)]·H2O}n (3). Complex 3 was synthesized by a procedure similar to that of 1, except that H2ppda (0.009 6 g, 0.05 mmol) replaced H2opda. Violet block crystals of 3 were obtained. Elemental analysis Calcd. for C22H20CoN4O5(%): C 55.12, H 4.21, N 11.69; Found(%): C 55.18, H 4.29, N 11.27.
1.3 Crystal structure determination
X-ray single-crystal diffraction data for complexes 1~3 were collected on a Bruker Smart 1000 CCD area-detector diffractometer with Mo Kα radiation (λ=0.071 073 nm) by ω scan mode. The crystal structure was solved by direct methods, using SHELXS-2014 and least-squares refined with SHELXL-2014[18]. Non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed in geometrically calculated positions. Further details for structural analysis are summarized in Table 1. Selected bond distances and bond angles are listed in Table S1.
Table 1

 下载:
导出CSV
下载:
导出CSV
Complex 1 2 3 Empirical formula C78H76Co3N16O18 C44H40Co2N8O10 C22H20CoN4O5 Formula weight 1 702.33 958.70 479.35 Crystal system Triclinic Monoclinic Orthorhombic Space group P1 P21/n Pna21 a/nm 1.100 0(6) 0.798 5(3) 1.889 4(2) b/nm 1.274 1(7) 3.794 6(14) 0.789 1(9) c/nm 1.534 4(8) 1.391 5(5) 1.429 1(16) α/(°) 103.410 0(10) β/(°) 108.677 0(10) 96.276 0(10) γ/(°) 95.755 0(10) V/nm3 1.945 8(18) 4.190 7(3) 2.131 3(4) Z 1 4 4 Dc/(g·cm-3) 1.453 1.519 1.494 μ/mm-1 0.713 0.862 0.848 Rint 0.010 5 0.117 6 0.034 3 Goodness-of-fit on F 2 1.194 1.034 1.044 R1a, wR2b [I>2σ(I)] 0.049 5, 0.132 3 0.061 3, 0.092 8 0.030 9, 0.069 6 R1a, wR2b (all data) 0.046 7, 0.131 0 0.149 7, 0.111 9 0.039 4, 0.073 0 a R1=∑||Fo|-|Fc||/∑|Fo|; b wR2=[∑w(Fo2-Fc2)2/∑w(Fo2)2]1/2 CCDC: 1856259, 1; 1856260, 2; 1856261, 3.
2. Results and discussion
2.1 Description of crystal structure
2.1.1 Crystal structure of {[Co1.5(opda)1.5(mbib)2(H2O)2]·H2O}n (1)
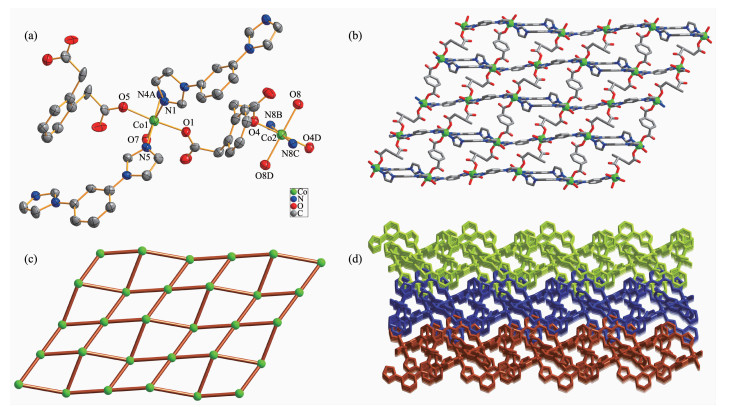
Single-crystal X-ray diffraction analysis reveals that complex 1 crystallizes in the triclinic system, P1 space group, and features a 2D network. The asym-metric unit of 1 consists of one and a half of cobalt(Ⅱ) ions, one and a half of fully deprotonated opda2- ligands, two mbib ligands, two coordinated water molecules and one lattice water molecule. The central Co1 atom has a distorted octahedral coordination geometry, coordinated by three nitrogen atoms of three different mbib ligands (Co1-N1 0.208 3(3) nm, Co1-N4A 0.208 1(4) nm, Co1-N5 0.206 8(4) nm) and three oxygen atoms from two different opda ligands and one water molecule (Co1-O1 0.205 6(3) nm, Co1-O5 0.207 4(3) nm, Co1-O7 0.212 7(3) nm). While the Co2 central atom is six-coordinated with a distorted octahedral coordination sphere, which is formed by two nitrogen atoms of two different mbib ligands (Co2-N8B/N8C 0.206 6(4) nm) and four oxygen atoms from three different opda2- ligands and one water molecule (Co2-O4/O4D 0.206 9(3) nm, Co2-O8/O8D 0.210 1(3) nm) (Fig. 1a).
Figure 1
 Figure 1. (a) Coordination environments of Co(Ⅱ) ion in complex 1 with 30% probability displacement ellipsoids; (b) Ball-and-stick view of 2D layer in 1; (c) Schematic view of (4, 4) net in 1; (d) View of 3D network of 1 extended by 2D layers in an offset stacking fashion
Figure 1. (a) Coordination environments of Co(Ⅱ) ion in complex 1 with 30% probability displacement ellipsoids; (b) Ball-and-stick view of 2D layer in 1; (c) Schematic view of (4, 4) net in 1; (d) View of 3D network of 1 extended by 2D layers in an offset stacking fashionAll hydrogen atoms are omitted for clarity; Symmetry codes: A: 1-x, -y, -z; B: 2-x, 1-y, 1-z; C:-1+x, -1+y, +z; D: 1-x, -y, 1-z
In the structure of 1, there are two kinds of mbib ligand showing non-planar conformation with the dihedral angle between benzene core and imidazole arms being 69.00° and 9.04°, 23.09° and 4.88°, respectively. Thus, the two unique Co1 atoms are bridged by two mbib ligands to form a paddle-wheel [(Co1)2(mbib)2] loop. Adjacent ones are further conn-ected through [Co2(mbib)2] motif to generate a 1D loop containing polymer chain (Fig.S1). The chains are further extended by opda2- in μ1-η1:η0 bis(monodentate) coordination modes to form a 2D (4, 4) net (Fig. 1b and 1c). In addition, viewed along c direction, the above-mentioned adjacent 1D chains are further interlinked to extend into an overall 3D hydrogen-bonded network (Fig. 1d) by the co-effects of the inter-layer C-H…π (C25-H25…π) stacking between benzene rings of mbib and opda2- ligands in an edge-to-face orientation with the C…π separations of 0.356 4 nm as well as C-H…O hydrogen-bonding interactions between benzene rings and carboxylate O atoms of opda2- ligands (C24-H24…O5).
2.1.2 Crystal structure of {[Co(mpda)(mbib)]·H2O}n (2)
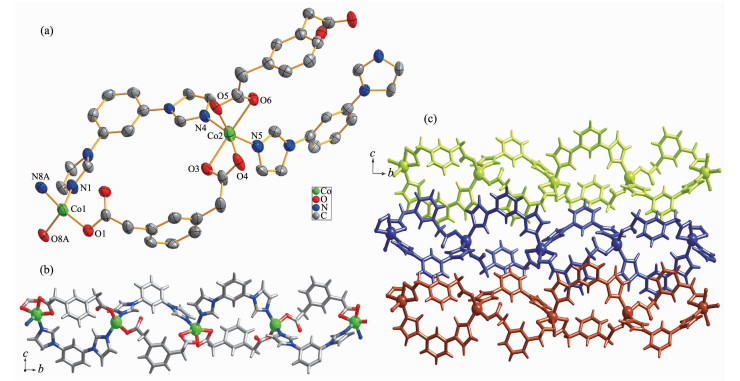
Utilizing the analogous bridging ligand mpda2-, instead of the opda2-, the 1D polymer chain of 2 was obtained. Single-crystal X-ray diffraction analysis reveals that complex 2 crystallizes in the monoclinic system, P21/n space group, and the asymmetric unit of 2 contains two crystallographically independent Co(Ⅱ) centers, each having a different coordination sphere. As shown in Fig. 2a, Co1 centers adopt slightly distorted tetrahedral geometries, coordinated by two nitrogen atoms of two mbib ligands (Co1-N1 0.204 0(2) nm, Co1-N8A 0.200 6(3) nm) and two oxygen atoms from two mpda2- ligands (Co1-O1 0.195 7(2) nm, Co1-O8A 0.195 5(2) nm). Co2 has a slightly distorted octahedral coordination environment, formed by four carboxyl oxygen atoms from two different mpda2- ligands(Co2-O3 0.218 1(2) nm, Co2-O4 0.225(3) nm, Co2-O5 0.190 1(18) nm, Co2-O6 0.225 5(2) nm) and two nitrogen atoms from two different mbib molecules (Co2-N4 0.206 0(2)nm, Co2-N5 0.208 1(3) nm).
Figure 2
 Figure 2. (a) Coordination environments of Co(Ⅱ) ion in complex 2 with 30% probability displacement ellipsoids; (b) Ball-and-stick view of 1D loop-containing chain along b direction in 2; (c) View of 2D extended supramolecular layer in 2
Figure 2. (a) Coordination environments of Co(Ⅱ) ion in complex 2 with 30% probability displacement ellipsoids; (b) Ball-and-stick view of 1D loop-containing chain along b direction in 2; (c) View of 2D extended supramolecular layer in 2All hydrogen atoms are omitted for clarity; Symmetry codes: A:-x+1/2, y-1/2, -z+1/2
As same as 1, the mbib ligands in the structure of 2 also exhibit non-planar conformation with the dihedral angle between benzene core and imidazole arms of 37.36° and 15.85°, 37.12° and 37.97°, respectively. In 1, the opda2- ligand has a trans-conformation with two carboxyl groups locating in two sides of the benzene ring plane, which can be viewed as a linear building block. Differently, the mpda2- ligand in 2 shows a cis-conformation with two carboxyl groups lying on the same sides of the benzene ring plane and acts as a "V" type building block. Thus, two kinds of such "V" type building block link the Co(Ⅱ) ions giving rise to a 1D loop containing polymeric ribbon running along b direction (Fig. 2b). In addition, the adjacent ribbon motifs are arranged into a 2D plane parallel to the bc plane by the co-effects of the inter-layer C-H…π (C43-H43…π) stacking between benzene ring of mbib and methyl of mpda2- ligand in an edge-to-face orientation with the C…π separations of 0.407 3 nm as well as C-H…O hydrogen-bonding interactions between imidazole rings of mbib and carboxylate O atoms of mpda2- ligands (C24-H24…O1, C24…O1 0.321 5 nm).
2.1.3 Crystal structure of {[Co(ppda)(mbib)]·H2O}n (3)
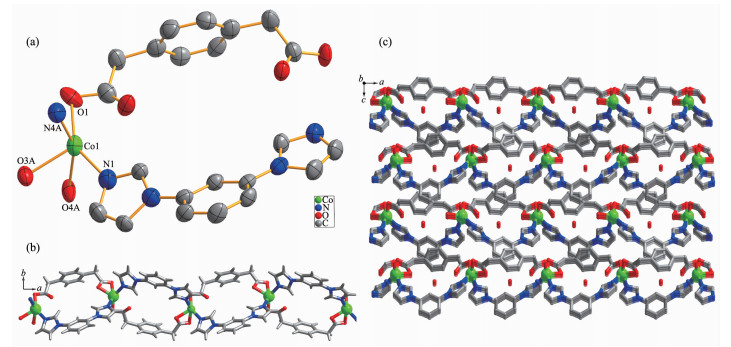
When the ppda2- building block was used, the similar 1D polymeric ribbon of 3 can also be isolated. Single crystal X-ray structure analysis reveals that the asymmetric unit of 3 contains one Co(Ⅱ) ion, one fully deprotonated ppda2- ligand, one mbib ligand and one lattice water molecule. As shown in Fig. 3a, unlike in 1 and 2, each Co(Ⅱ) ion in 3 is penta-coordinated by two N-atom donors from two mbib ligands (Co1-N1 0.205 2(3) nm, Co1-N4A 0.204 7(3) nm) and three O atoms from two different ppda2- ligands (Co1-O1 0.220 4(18) nm, Co1-O3A 0.212 8(3) nm, Co1-O4A 0.204 7(3) nm).
Figure 3
 Figure 3. All hydrogen atoms are omitted for clarity; Symmetry code: A: x-1/2, -y+3/2, z
Figure 3. All hydrogen atoms are omitted for clarity; Symmetry code: A: x-1/2, -y+3/2, z(a) Coordination environments of Co(Ⅱ) ion in complex 3 with 30% probability displacement ellipsoids; (b) Ball-and-stick view of 1D loop-containing chain along a direction in 3; (c) View of 3D porous supramolecular network in 3 with lattice molecules in the channel
In the structure of 3, the dihedral angle between two imidazole arms and benzene core is 32.91° and 14.22°, respectively. Two carboxyl groups of ppda2- in 3 adopt monodentate and chelate coordination mode and point to the same side of the benzene ring plane. Thus, the ppda2- ligand also has a cis-conformation and can be seen as a "V" type building block. Co(Ⅱ)ions are joined together to form a 1D loop containing polymeric ribbon running along a direction (Fig. 3b) by the mixture of two "V" type building block: mbib and ppda2- ligands. The Co…Co separation through mbib ligand in 3 (0.957 5 nm) is close to those in 2 (0.950 3 and 0.969 3 nm) and shorter than that in 1 (1.090 4 nm). The crystal packing diagram of 3 shows that the adjacent 1D polymeric ribbon motif are bound together by strong intermolecular hydrogen bonds to create a 2D supramolecular layer (Fig.S2). The hydrogen bond-ing system in 3 consists of the C-H…O (C2…O4 0.363 2 nm, ∠C2-H2B…O4=163.18°) hydrogen bond-ing interactions between methyl of ppda2- from ribbon and carboxylate O atoms from the other ppda2-. In addition, the extended 3D porous supramolecular network (Fig. 3c) is formed through π…π interac- tions (centroid-centroid distance 0.397 5 nm) between benzene rings of ppda2- and mbib ligands with a face-to-face orientation. The lattice water molecules are fixed in the channels through C-H…O (C13…O5 0.348 4 nm, ∠C13-H13…O5=170.49°; C17…O5 0.336 1 nm, ∠C17-H17…O5=163.1843°; C23…O5 0.352 6 nm, ∠C23-H123…O5=153.95°) and O-H…O (O5…O2 0.294 4 nm, ∠O5-H5A…2=151.73°; O5…O3 0.338 3 nm, ∠O5-H5B…O3=146.26°) hydrogen bonding interactions. There is no doubt that these strong hydrogen-bonding interactions play an important role in the formation of the 3D supramolecular architecture.
2.2 PXRD results and thermal analysis of complexes 1~3
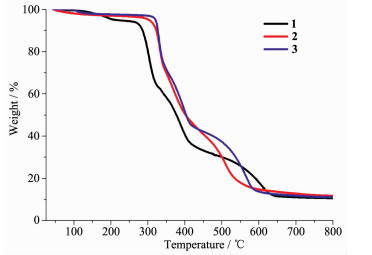
In order to confirm the phase purity of the bulk materials, powder X-ray diffraction (PXRD) patterns of complexes 1~3 were recorded at room temperature. As shown in Fig.S3, the peak positions of the experimental PXRD and computer-simulated patterns are in agreement with each other, which confirms its phase purity. The difference in intensity of some diffraction peaks may be attributed to the preferred orientation of the crystalline powder samples. To examine the stability of these complexes, thermal gravimetric analysis (TGA) experiment was performed. As shown in Fig. 4, the TGA curves exhibit a weight loss of 5.9% (95~208 ℃) for 1, 3.3% (80~130 ℃) for 2, and 3.5% (78~135 ℃) for 3, corresponding to the release of coordinated water molecules and/or free water molecules (Calcd. 6.1%, 3.7%, and 3.7% for 1~3, respectively). The skeleton of 1~3 can be stable up to 267, 300 and 311 ℃, respectively.
Figure 4
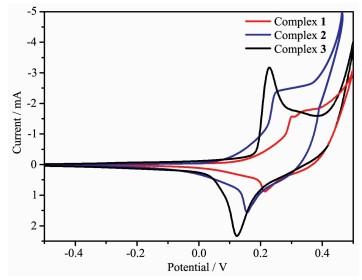
2.3 Electrochemistry properties
In the cyclic voltammetry (CV) measurement of complexes, we employed a conventional three-electrodes system where a saturated calomel electrode (SCE) as the reference electrode, platinum electrodes as auxiliary electrode, and foam nickel electrode was chosen as the working electrode. Water was used as solvent, and the supporting electrolyte was 1 mol·L-1 KOH solution. Cyclic voltammograms of complexes 1~3 are shown in Fig. 5. During scanning from -0.5 to 0.5 V in a rate of 100 mV·s-1, the cyclic voltammogram curves had an oxidation-reduction peak, which corres-ponds to Co(Ⅱ)/Co(Ⅰ) redox process[19-21]. The cyclic voltammograms of complexes 1, 2 and 3 all showed a reversible redox wave, and the observed potential were as follows: E1/2=0.258 V (ΔEp=0.089 V) for 1, E1/2=0.202 V (ΔEp=0.091 V) for 2, E1/2=0.178 V (ΔEp=0.105V) for 3. For complexes 1~3, their electro-chemical behaviors are similar except for some slight potential shifts, due to the similarity of the different ligands that make up the three complexes.
Figure 5
3. Conclusions
In summary, three new coordination polymers {[Co1.5(opda)1.5(mbib)2(H2O)2]·H2O}n (1), {[Co(mpda)(mbib)]·H2O}n (2) and {[Co(ppda)(mbib)]·H2O}n (3) have been synthesized under hydrothermal conditions by the reaction of Co(Ⅱ) salt and bis(imidazole) ligand in the presence of different flexible phenylenedia-cetate blocks. In these complexes, the central Co(Ⅱ) ions show different coordination geometries, the opda2- ligand exhibits trans-conformation and acts as bridging building block to extend 1D loop containing (Co-mbib) polymer chain into a 2D network in 1. While mpda2- and ppda2- ligands in 2 and 3 possesses cis-conforma-tion and can be seen as "V" type building block, linking Co(Ⅱ) ions to form 1D loop containing chain together with mbib ligand. The structural diversities of these complexes are significantly affected by the coordination geometries of the central metal ions and the coordination modes, conformations of the organic ligands. All of three complexes are electrochemically active. This study is significant for extending the metal-organic complexes containing electronic conjugated system, exploring the interrelation between structure and property to develop potential electrochemical functional materials.
Supporting information is available at http://www.wjhxxb.cn
-
-
[1]
Li D S, Wu Y P, Zhao J, et al. Coord. Chem. Rev., 2014, 261:1-27 doi: 10.1016/j.ccr.2013.11.004
-
[2]
Zhao Y, Deng D S, Ma L F, et al. Chem. Commun., 2013, 49:10299-10301 doi: 10.1039/c3cc45310c
-
[3]
Hu Z, Deibert B J, Li J. Chem. Soc. Rev., 2014, 43:5815-5840 doi: 10.1039/C4CS00010B
-
[4]
Li C P, Wang S, Guo W, et al. Chem. Commun., 2016, 52:11060-11063 doi: 10.1039/C6CC05938D
-
[5]
Yang X G, Yan D P. Adv. Opt. Mater., 2016, 4:897-905 doi: 10.1002/adom.201500666
-
[6]
Li B, Wen H M, Cui Y, et al. Adv. Mater., 2016, 28:8819-8860 doi: 10.1002/adma.201601133
-
[7]
Zhang Y, Yuan S, Day G, et al. Coord. Chem. Rev., 2018, 354:28-45 doi: 10.1016/j.ccr.2017.06.007
-
[8]
Cui Y, Yue Y, Qian G, et al. Chem. Rev., 2011, 112:1126-1162
-
[9]
Yu Y, Yue C, Lin X, et al. ACS Appl. Mater. Interfaces, 2016, 8:3992-3999 doi: 10.1021/acsami.5b11287
-
[10]
Horcajada P, Gref R, Baati T, et al. Chem. Rev., 2012, 112:1232-1268 doi: 10.1021/cr200256v
-
[11]
Zheng H, Zhang Y, Liu L, et al. J. Am. Chem. Soc., 2016, 138:962-968 doi: 10.1021/jacs.5b11720
-
[12]
Fan J, Sun W Y, Okamura T, et al. Inorg. Chem., 2003, 42:3168-3175 doi: 10.1021/ic0206847
-
[13]
Ma L F, Wang Y Y, Liu J Q, et al. CrystEngComm, 2009, 11:1800-1802 doi: 10.1039/b904977k
-
[14]
Su K, Jiang F L, Qian J J, et al. Inorg. Chem., 2013, 52:3780-3786 doi: 10.1021/ic302367q
-
[15]
Luo J H, Jiang F L, Wang R H, et al. Inorg. Chem. Commun., 2004, 7:638-642 doi: 10.1016/j.inoche.2004.03.007
-
[16]
Zhang M L, Wang J J, Ma Z Z, et al. New J. Chem., 2017, 41:12139-12146 doi: 10.1039/C7NJ00645D
-
[17]
Zhang M L, Zheng Y J, Ma Z Z, et al. Polyhedron, 2018, 146:180-186 doi: 10.1016/j.poly.2018.03.005
-
[18]
Sheldrick G M. Acta Crystallogr. Sect. A:Found. Crystallogr., 2008, 64:112-122 doi: 10.1107/S0108767307043930
-
[19]
Deilami A B, Salehi M, Arab A. Inorg. Chim. Acta, 2018, 476:93-100 doi: 10.1016/j.ica.2018.02.013
-
[20]
Lim I T, Choi K Y. Molecules, 2013, 18:6608-6619 doi: 10.3390/molecules18066608
-
[21]
王庆华, 于丽丽, 刘琦, 等.无机化学学报, 2011, 27:989-995 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20110531&flag=1WANG Qin-Hua, YU Li-Li, LIU Qi, et al. Chinese J. Inorg. Chem., 2011, 27:989-995 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20110531&flag=1
-
[1]
-
Figure 1 (a) Coordination environments of Co(Ⅱ) ion in complex 1 with 30% probability displacement ellipsoids; (b) Ball-and-stick view of 2D layer in 1; (c) Schematic view of (4, 4) net in 1; (d) View of 3D network of 1 extended by 2D layers in an offset stacking fashion
All hydrogen atoms are omitted for clarity; Symmetry codes: A: 1-x, -y, -z; B: 2-x, 1-y, 1-z; C:-1+x, -1+y, +z; D: 1-x, -y, 1-z
Figure 2 (a) Coordination environments of Co(Ⅱ) ion in complex 2 with 30% probability displacement ellipsoids; (b) Ball-and-stick view of 1D loop-containing chain along b direction in 2; (c) View of 2D extended supramolecular layer in 2
All hydrogen atoms are omitted for clarity; Symmetry codes: A:-x+1/2, y-1/2, -z+1/2
Figure 3 All hydrogen atoms are omitted for clarity; Symmetry code: A: x-1/2, -y+3/2, z
(a) Coordination environments of Co(Ⅱ) ion in complex 3 with 30% probability displacement ellipsoids; (b) Ball-and-stick view of 1D loop-containing chain along a direction in 3; (c) View of 3D porous supramolecular network in 3 with lattice molecules in the channel
Table 1. Crystallographic data and refinement parameters for complexes 1~3
Complex 1 2 3 Empirical formula C78H76Co3N16O18 C44H40Co2N8O10 C22H20CoN4O5 Formula weight 1 702.33 958.70 479.35 Crystal system Triclinic Monoclinic Orthorhombic Space group P1 P21/n Pna21 a/nm 1.100 0(6) 0.798 5(3) 1.889 4(2) b/nm 1.274 1(7) 3.794 6(14) 0.789 1(9) c/nm 1.534 4(8) 1.391 5(5) 1.429 1(16) α/(°) 103.410 0(10) β/(°) 108.677 0(10) 96.276 0(10) γ/(°) 95.755 0(10) V/nm3 1.945 8(18) 4.190 7(3) 2.131 3(4) Z 1 4 4 Dc/(g·cm-3) 1.453 1.519 1.494 μ/mm-1 0.713 0.862 0.848 Rint 0.010 5 0.117 6 0.034 3 Goodness-of-fit on F 2 1.194 1.034 1.044 R1a, wR2b [I>2σ(I)] 0.049 5, 0.132 3 0.061 3, 0.092 8 0.030 9, 0.069 6 R1a, wR2b (all data) 0.046 7, 0.131 0 0.149 7, 0.111 9 0.039 4, 0.073 0 a R1=∑||Fo|-|Fc||/∑|Fo|; b wR2=[∑w(Fo2-Fc2)2/∑w(Fo2)2]1/2  下载: 导出CSV
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 1056
- HTML全文浏览量: 144
