Table 1.
Optimization of reaction conditionsa
Citation:
Li Pengshuai, Wu Yun, Bai Chaolumen, Bao Yongsheng. Insight into Catalytic Properties of Supported Palladium Nanoparticles Catalyzed ortho-Directed Sulfonylation[J]. Chinese Journal of Organic Chemistry,
2020, 40(7): 1991-1998.
doi:
10.6023/cjoc202003020

负载纳米钯颗粒在邻位导向磺化反应中的催化特性探究
English
Insight into Catalytic Properties of Supported Palladium Nanoparticles Catalyzed ortho-Directed Sulfonylation
Abstract:
Catalyzed by supported palladium nanoparticles, an ortho-directed sulfonylation reaction between 2-phenylpyri-dine and arylsulfonyl chlorides has been developed. The full oxidation-state change of palladium was detected in the X-ray photoelectron spectroscopy (XPS) analysis of the supported palladium nanoparticles catalyst before and after reaction, which confirmed that Pd-catalyzed ortho-directed sulfonylation reaction was performed via a PdⅡ/IV catalytic cycle instead of Pd0/Ⅱ. The hot filtration test and other tests of catalysts further confirmed the hypothesis. This report afforded the most straightforward approach to confirm the variation of valence of palladium in ortho-directed sulfonylation reaction.
-
1. Introduction
Compared with the traditional cross coupling methods, the C—H activation/functionalization approach avoids the use of prefunctionalized substrates, and thereby provides means for environmentally-benign and economically attractive organic syntheses.[1] Palladium complexes are particularly attractive catalysts for such transformations because that C—H functionalization at Pd centres can be used to install many different types of bonds and palladium participates in cyclometalation with a wide variety of directing groups and readily promotes C—H activation at both sp2- and sp3-C—H sites.[2] But one can question whether the C—H functionalization reaction mechanisms involving Pd are fully understood. To date the mechanistic picture is dominated by homogeneous catalytic cycles involving one or two Pd atoms, involving Pd0, PdII, PdIII and PdIV oxidation states.[3] The directed palladium catalyzed oxidative functionalizations of C—H bonds are typically believed to proceed through PdII/PdIV redox cycles.[4] Ritter et al.[5] proposed a challenging mechanism for C—Cl, C—B and
C—O reductive eliminations from discrete bimetallic PdIII complexes. By contrast, it was proposed that the majority of palladium catalyzed C—H activation reactions do in fact proceed through a Pd0/II manifold, [6] because the propagation of Pd black, Pd mirror or PdNPs from common Pd precursors (Pd(Oac)2) was found in many reactions.[7] But sometimes the mechanism understanding of the same palladium catalyzed C—H activation reactions is often different in proceeding Pd0/II or PdII/IV process because of the difficulty of isolating the Pd intermediates in the homogeneous catalytic system.[8] It is believed that this has become one of the most intriguing open problems in homogeneous catalysis.
In recent years, pre-supported palladium nanoparticle (PdNP) catalysts have attracted broad interest of chemists due to their high selectivity and efficiency as the heterogeneous catalyst for various reactions, such as hydrogenation of conjugated dienes, enantioselective allylic alkylation, carbon-carbon coupling, asymmetric allylic substitution, and electrocatalytic formic acid oxidation.[9] Compared with homogeneous catalysts, the easy separation and reusability of heterogeneous catalysts would be desirable to develop novel and efficient approach for mechanism study. The most straightforward approach to confirm the valence states of palladium in reaction is the X-ray photoelectron spectroscopy (XPS) analysis of the catalyst.[10] XPS is sensitive to small changes in the chemical environment and thus is the ideal tool to verify the effect of functionalization on the surface chemistry of Pd nanoclusters.[11] In heterogeneous catalysis system, the beginning and ending of catalytic cycle could be confirmed by the XPS analysis of the catalyst before and after reaction (commonly after multiple cycles).[12] In 2011, Qi and co-workers[13] used the XPS studies to speculate that the Pd/SP catalyzed reductive homocoupling of aromatic halides in ethanol/dimethyl sulfoxide (DMSO) solution proceeds through a Pd0/PdII cycle catalytic mechanism. Xing and co-worker[14] reported an effective redox cycle between Pd0 and PdII in the catalytic hydrochlorination of acetylene using the XPS studies. But, as far as we know, there is rare report on using the XPS studies to research the change of valence state of palladium in C—H activation reaction.
Sulfones have been extensively proven to be vital intermediates in organic synthesis, because of unique chemical properties and bioactivities, such as anticancer, anti-HIV and antibacterial.[15] In 2009, Dong and co-workers[8b] reported that Pd(CH3CN)2Cl2 catalyzed the synthesis of ortho-aromatic sulfones using arylsulfonyl chlorides as sulfonyl sources. But the indicated C—S bond-forming process involving Pd0/II or PdII/IV intermediates was not proposed explicitly. In 2015, Sun et al.[16] reported that Pd(Oac)2 catalyzed the ortho-sulfonylation of 2-aryloxypyridines and arylsulfonyl chlorides. They proposed the PdII/IV mechanism according to the references instead of other direct evidences.
Based on our researches on the supported PdNPs catalyzed C—H activation reaction, we wished to gain some insight into catalytic properties of palladium catalyzed ortho-directed sulfonylation by means of XPS analysis. The XPS analysis of the supported palladium nanoparticle catalyst before and after reaction confirmed that there was not a redox cycle between Pd0 and PdII. The heterogeneity test and extensive Pd leaching suggested that the supported PdNPs catalyzed ortho-directed sulfonylation should proceed in a homogeneous catalytic cycle that began with PdII.
2. Results and discussion
The activity of the Pd/γ-al2O3 catalyst was first investigated by the ortho-directed sulfonylation of 2-phenylpyri- dine (1a) with 4-methoxy-benzenesulfonyl chloride (2a) in ethylbenzene and the desired product 3aa was isolated in 73% yield (Table 1, Entry 1). The catalytic activity of PdNPs was slightly lower than that of Pd(Oac)2 (Table 1, Entry 2). The reaction proceeded smoothly over Pd/NiO (wPd=3%), Pd/TiO2 (wPd=3%) and Pd/ZrO2 (wPd=3%) catalysts, while the obtained yields were much lower than that over Pd/γ-al2O3 (wPd=3%) catalyst (Table 1, Entries 3~5). Moreover, the effect of different Pd loadings on the reaction was also examined, and Pd/γ-al2O3 (wPd=3%) was the most effective catalyst (Table 1, Entries 6, 7). It was noteworthy that no reaction was observed in a blank experiment conducted with γ-al2O3 powder instead of PdNPs under otherwise identical conditions (Table 1, Entry 8). Both an increase and decrease in the reaction temperature reduced the yield of 2-(2-(4-methoxylphenylsulfonyl)phe- nyl)pyridine (3aa) (Table 1, Entries 10, 11).
Table 1

 下载:
导出CSV
下载:
导出CSV

Entry Catalyst Temp./℃ Yield/% 1 Pd/γ-al2O3 (wPd=3%) 125 73 2 Pd(Oac)2 125 75 3 Pd/NiO (wPd=3%) 125 33 4 Pd/TiO2 (wPd=3%) 125 52 5 Pd/ZrO2 (wPd=3%) 125 56 6 Pd/γ-al2O3 (wPd=1%) 125 54 7 Pd/γ-al2O3 (wPd=5%) 125 61 8 γ-al2O3 125 0 10 Pd/γ-al2O3 (wPd=3%) 130 70 11 Pd/γ-al2O3 (wPd=3%) 115 65 a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), catalyst (35 mg), K2CO3 (0.4 mmol), and ethylbenzene (2 mL) at 125 ℃ for 24 h. b Isolated yield. The scope of the method of synthesizing a broad range of sulfones was investigated with a variety of arylsulfonyl chlorides and 2-phenylpyridine derivatives under the optimized reaction conditions, and the results were shown in Table 2. The initial investigations were focused on the ortho-directed sulfonylation of 2-phenylpyridine (1a) with various arylsulfonyl chlorides (Table 2, Entries 1~8). To our delight, various substituted arylsulfonyl chlorides were successfully converted to the corresponding sulfones in moderate yields. Our experiment indicated that the arylsulfonyl chlorides bearing electron-withdrawing groups on the aromatic ring gave lower yields than others and 4-nitro- benzenesulfonyl chloride (2e) led to the lowest yield of the corresponding sulfone (3ae). 4-tert-Butylbenzene-sulfonyl chloride (2d) which bears an electron-donating group afforded the desired product 2-(2-(4-tert-butylphenylsulfo- nyl)-phenyl)-pyridine (3ad) in the best yield of 78% (Table 2, Entry 3). In addition, 2-naphthalenesulfonyl chloride (2f) and 1-naphthalenesulfonyl chloride (2g) as well as benzenesulfonyl chloride performed the sulfonylation reaction to give the corresponding sulfones in moderate yields (Table 2, Entries 5, 6). apart from aromatic sulfonyl chloride, the methodology was also successful in converting heteroaromatic sulfonyl chloride to the corresponding products (Table 2, Entries 7, 8). The sulfonylation of 2-(p-tolyl)pyridine (1b) with 4-tert-butylbenzenesulfonyl chloride (2d) were investigated, and the desired product were isolated in moderate yields (Table 2, Entry 9). Using 1-phenyl-1H-pyrazole (1c) as a substrate resulted in desired 1-(2-(4-tert-butyl- phenylsulfonyl)phenyl)-1H-pyrazole (3cd) albeit in comparatively lower yield (Table 2, Entry 10).
Table 2
Table 2. Substrate scope of ortho-directed sulfonylationa下载:
导出CSV

Entry 1 2 Product Yieldb/% Entry 1 2 Product Yieldb/% 1 1a 

51 6 1a 

29 2 1a 

48 7 1a 

55 3 1a 

78 8 1a 

57 4 1a 

18 9 1b 

73 5 1a 

43 10 1c 

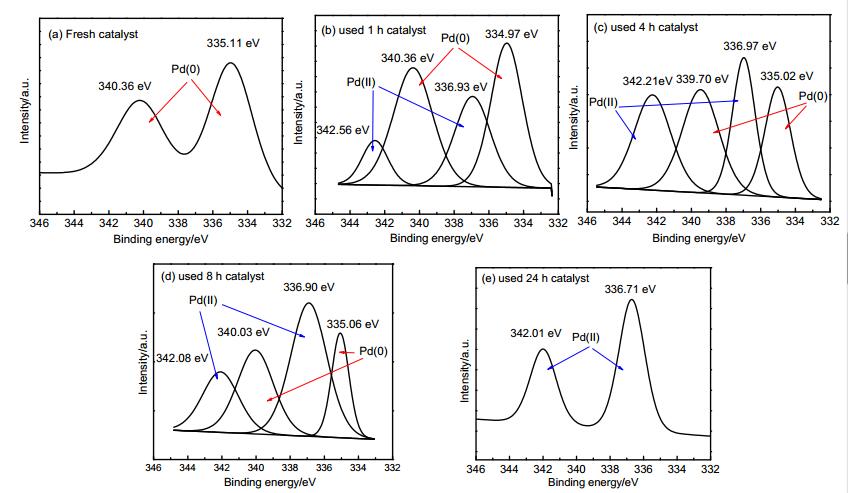
28 a Reaction conditions: 1 (0.2 mmol), 2 (0.6 mmol), Pd/γ-al2O3 (wPd=3%, 35 mg), K2CO3 (0.4 mmol), ethylbenzene (2 mL), 125 ℃, 24 h. b Isolated yield. In order to get information on the valence state of Pd on the surface of the catalyst, the fresh and used (after 1st cycle) Pd/γ-al2O3 (wPd=3%) catalysts were tested by XPS analysis (Figure 1a, 1e). The XPS signal appeared at binding energies for 335.11 eV of Pd 3d5/2 and 340.36 eV of Pd 3d3/2 over fresh catalyst and 336.71 eV of Pd 3d5/2 and 342.01 eV of Pd 3d3/2 over used catalyst. Obviously, the Pd 3d3/2 binding energy observed in the XPS spectrum of the Pd/al2O3 is shifted by ca +1.60 eV revealing a full oxidation of the Pd atoms. The binding energies of Pd0 in literature are 335.20 eV of Pd 3d5/2 and 340.50 eV of Pd 3d3/2, re-spectively. While the values of PdII are 336.70 eV of Pd 3d5/2 and 342.00 eV of Pd 3d3/2, respectively.[17] These results showed that the supported metallic PdNPs were fully oxidized to divalent Pd species after reaction 24 h. aim to get more detail process about the full oxidation of the Pd atoms, the catalysts after reaction for different times were isolated and detected by XPS. as shown in Figure 1, the co-existence of Pd0 and PdII was observed in the XPS spectra of catalysts after reaction 1, 4 and 8 h. However, according to the area integral, the fraction of PdII in total metal species increased continuously in the reaction progress and after reacting 8 h PdII had occupied a dominant position. It suggested that Pd0 could continuously be oxidized to PdII, but PdII was not reduced to Pd0 in the reaction process. after reacting 24 h, Pd0 disappeared. Meanwhile, the corresponding continuously changes of catalyst colours were also detected (Figure 2). This was opposite to the XPS study result of Pd/γ-al2O3 catalyzed ortho-directed CDC reaction in our previous work.[18] Hence, it is fairly probable that there is not a redox cycle between Pd0 and PdII in the ortho-directed sulfonylation reaction and PdII may be served as catalytic centre.
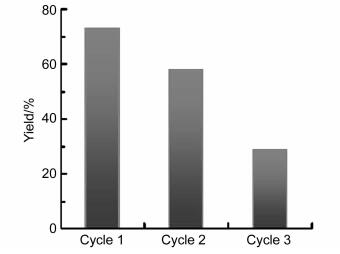
Then, the possibility of recycling the oxidized PdNP catalyst in the model reaction was examined (Figure 3). The divalent supported PdNPs could recycled 3 times, although the catalytic performance of the PdNPs declined apparently after every once recycle. This further confirmed the hypothesis that the Pd-catalyzed ortho-directed sulfonylation reaction was performed via a catalytic cycle that began with PdII instead of Pd0.
Figure 1
 Figure 1. XPS spectra of fresh and used Pd/γ-al2O3 (wPd=3%) (after reaction different times)
Figure 1. XPS spectra of fresh and used Pd/γ-al2O3 (wPd=3%) (after reaction different times)Figure 2
 Figure 2. Photos of fresh and used Pd/γ-al2O3 (wPd=3%) (after reaction different times)
Figure 2. Photos of fresh and used Pd/γ-al2O3 (wPd=3%) (after reaction different times)Figure 3
spectively. While the values of PdII are 336.70 eV of Pd 3d5/2 and 342.00 eV of Pd 3d3/2, respectively.[17] These results showed that the supported metallic PdNPs were fully oxidized to divalent Pd species after reaction 24 h. aim to get more detail process about the full oxidation of the Pd atoms, the catalysts after reaction for different times were isolated and detected by XPS. as shown in Figure 1, the co-existence of Pd0 and PdII was observed in the XPS spectra of catalysts after reaction 1, 4 and 8 h. However, according to the area integral, the fraction of PdII in total metal species increased continuously in the reaction progress and after reacting 8 h PdII had occupied a dominant position. It suggested that Pd0 could continuously be oxidized to PdII, but PdII was not reduced to Pd0 in the reaction process. after reacting 24 h, Pd0 disappeared. Meanwhile, the corresponding continuously changes of catalyst colours were also detected (Figure 2). This was opposite to the XPS study result of Pd/γ-al2O3 catalyzed ortho-directed CDC reaction in our previous work.[18] Hence, it is fairly probable that there is not a redox cycle between Pd0 and PdII in the ortho-directed sulfonylation reaction and PdII may be served as catalytic centre.
Then, the possibility of recycling the oxidized PdNP catalyst in the model reaction was examined (Figure 3). The divalent supported PdNPs could recycled 3 times, although the catalytic performance of the PdNPs declined apparently after every once recycle. This further confirmed the hypothesis that the Pd-catalyzed ortho-directed sulfonylation reaction was performed via a catalytic cycle that began with PdII instead of Pd0.
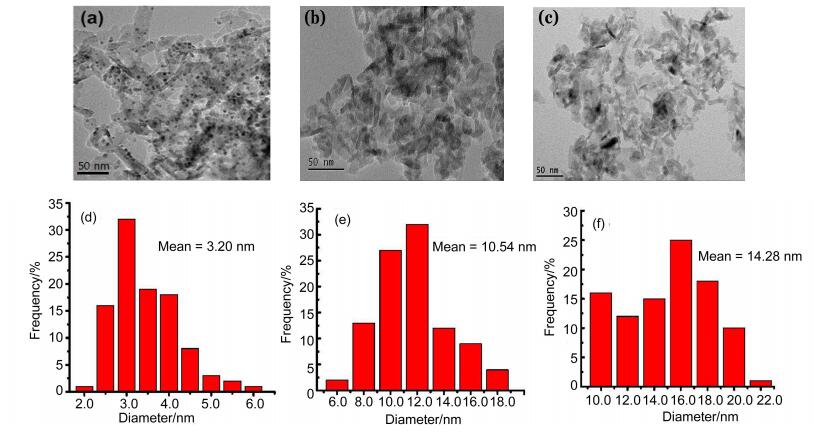
The dispersed states of Pd0 and PdII NPs on the support were detected by the transmission electron microscopic (TEM). as shown in Figure 4, the Pd0 NPs are distributed evenly on the γ-al2O3 surface, and the mean diameters of the PdNPs are 3.20 nm in fresh catalyst. However, after 1st cycle, larger aggregated particles with a mean particle size estimate as 10.54 nm was detected. Likewise, after 3th cycle, the aggregation of PdNPs was much more serious. This indicated that without Pd0 served as reservoirs, PdII hard to reassemble into small sized PdNPs on the supporter surface. The aggregation of metal NPs during the catalysis processes results in a significant decrease of acitivity.[19]
Figure 4
 Figure 4. TEM images of fresh (a), after 1th cycle (b) and after 3th cycle (c) Pd/γ-al2O3 (wPd=3%), and the PdNPs size distributions of fresh (error range: ±0.25 nm) (d), after 1th cycle (Error range: ±1.0 nm) (e) and after 3th cycle (Error range: ±1.0 nm) (f) catalysts, respectively
Figure 4. TEM images of fresh (a), after 1th cycle (b) and after 3th cycle (c) Pd/γ-al2O3 (wPd=3%), and the PdNPs size distributions of fresh (error range: ±0.25 nm) (d), after 1th cycle (Error range: ±1.0 nm) (e) and after 3th cycle (Error range: ±1.0 nm) (f) catalysts, respectivelyThe amounts of Pd loading in the samples were determined by atomic absorption spectrophotometer (aaS), and the Pd content of the fresh catalyst was approximately 3% (Table 3). We did note a continuously decrease in the Pd content after being cycles 3 times, which confirmed that there was the extensive Pd leaching of Pd/γ-al2O3 in the sulfonylation reaction.[20] apparently, it could decrease the catalytic activity on the basis of available Pd of the catalyst.
Table 3
Table 3. Characterization results of aaS of different Pd/γ-al2O3 catalysts下载:
导出CSV
Sample Pd loading w/% Pd leaching
rate/%3% Pd/γ-al2O3 (fresh) 3.01 3% Pd/γ-al2O3 (after 1th cycle) 2.36 -22 3% Pd/γ-al2O3 (after 3th cycle) 0.51 -83 In order to prove whether the present sulfonylation reaction proceeded with heterogeneous catalyst or homogeneous catalyst, the hot filtration test of the model reaction mixture was executed and GC data was given in Table 4. The conversion of 1a and turn over frequency (TOF) value indicated that after filtration the substrate turnover did not cease, instead the reaction can continuously proceed. This result indicated that oxidized PdII species, which served as catalytic active centre, extensively leached into solution from higher order aggregated Pd species, including Pd0 and PdII NPs, and the ortho-directed sulfonylation should pro- ceed in a homogeneous manner. Combined with the aaS results of fresh and used catalysts (Figure 1), we consider that the release of Pd is concomitant with recapture Pd at the catalyst surface, but releasing much more than recapturing.
Table 4
Table 4. The results of the hot filtration test下载:
导出CSV
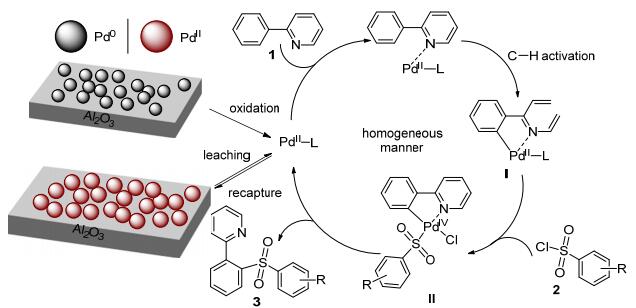
Time/h Conversiona/% TOF 0 0 2 27.9 2.81 4 (after hot filtration) 68.0 6.86 Based on the experiment results and references, the homogeneous manner manifold using PdII as active centre was proposed (Scheme 1). First, Pd0 NPs was oxidized to PdII species and leaching into reaction solution at reaction temperature. Simultaneously, PdII species was recaptured by supporter and aggregated to PdII NPs, so the fraction of PdII in total metal species increased continuously as the reaction progress. Instead of Pd0 NPs, the leached PdII species served as the true catalytic centre and performed the ortho-directed sulfonylation reaction by homogeneous manner. a PdIV intermediate II via oxidative addition of sulfonyl chloride to the cyclopalladated intermediate I was proposed in the palladium catalyzed ortho-directed sulfonylation.
Scheme1
3. Conclusion
In summary, the mechanism of supported palladium nanoparticles catalyzed ortho-directed sulfonylation has been researched by means of heterogeneous approach (the XPS analysis of catalysts). after reaction 24 h, the supported metallic PdNPs were fully oxidized to divalent Pd species. It confirms that there is not a redox cycle between Pd0 and PdII in the ortho-directed sulfonylation reaction. This is opposite to the XPS study result of Pd/γ-al2O3 catalyzed ortho-directed CDC reaction in our previous work. The experiment results indicated that the oxidized PdII species, which served as catalytic active centre, extensively leached into solution from higher order aggregated Pd species, including Pd0 and PdII NPs, and drive the ortho-directed sulfonylation reaction. The practicality of this heterogeneous approach for mechanism study may inspire further studies on the other palladium catalyzed C—H activation reaction.
4. Experimental
4.1 Catalyst preparation
The PdNPs on γ-al2O3 and other supports were prepared by a modified impregnation-reduction method.[18]
4.2 Catalyst characterization
The TEM images were recorded on a JEM-2100 transmission electron microscope employing an accelerating voltage of 200 kV. The samples were suspended in ethanol and dried on holey carbon-coated Cu grids. The X-ray photoelectron spectroscopy (XPS) was recorded on a Kratos amicus of British equipped with taper anode Mg Kα radiation. The C1s hydrocarbon peak at 284.60 eV was used as an internal standard for the correction of binding energies. The Pd content was determined by the Z-8000-type polarized Zeeman atomic absorption spectrophotometer (aaS) of Hitachi company. The au sensitive wavelength is 242.8 nm. GC-MS analysis was detected on a Thermo DSQ-II with a DB-5 column. Thin layer chromatography (TLC) was performed on pre-coated silica gel GF254 plates.
4.3 activity test
The reaction was conducted in a 25 mL of oven-dried reaction tube. 2-Phenylpyridine (1a) sulfonylation with 4-methoxy benzenesulfonyl chloride (2a) was used as the model reaction. In a typical reaction, catalyst (35 mg), 2-phenylpyridine (30 μL, 0.20 mmol), 4-methoxy benzenesulfonyl chloride (123.6 mg, 0.60 mmol), K2CO3 (2 equiv.) and ethylbenzene (2.0 mL) were added into the reac- tion tube. The resulting solution was stirred at 125 ℃ for 24 h in an oil bath. all the products were confirmed by comparison with the previously reported 1H NMR and 13C NMR data.
2-(2-(4-Methoxylphenylsulfonyl)-phenyl)-pyridine(3aa):[21] Yield 73% (47.5 mg). White solid, m.p. 128.2~129.8 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.42 (d, J=4.8 Hz, 1H), 8.31 (d, J=7.6 Hz, 1H), 7.75 (td, J=7.7, 1.5 Hz, 1H), 7.64~7.54 (m, 3H), 7.36 (dd, J=11.4, 5.1 Hz, 3H), 7.28 (d, J=8.5 Hz, 1H), 6.77 (d, J=8.9 Hz, 2H), 3.80 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 163.0, 156.8, 148.5, 140.0, 135.6, 132.9, 132.8, 132.0, 130.3, 130.0 (2C), 128.9, 128.7, 126.0, 122.6, 113.9 (2C), 55.6; MS (ESI) m/z: 326.05 [M+H]+.
2-(2-(4-Methylphenylsulfonyl)-phenyl)-pyridine (3ab):[22] Yield 51% (31.5 mg). White solid, m.p. 107 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.38 (d, J=4.3 Hz, 1H), 8.32 (d, J=7.5 Hz, 1H), 7.73 (t, J=7.3 Hz, 1H), 7.6~7.57 (m, 2H), 7.54 (d, J=7.8 Hz, 1H), 7.39~7.30 (m, 3H), 7.28~7.23 (m, 1H), 7.10 (d, J=8.0 Hz, 2H), 2.34 (s, 3H).
2-(2-Phenylsulfonylphenyl)-pyridine (3ac):[21] Yield 48% (28.3 mg). Slightly yellow solid, m.p. 90~91.6 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.35 (dd, J=7.6, 1.2 Hz, 2H), 7.72 (td, J=7.7, 1.7 Hz, 1H), 7.63 (m, J=15.1, 7.5, 1.5 Hz, 2H), 7.52 (d, J=7.8 Hz, 1H), 7.48~7.42 (m, 3H), 7.37 (dd, J=7.3, 1.4 Hz, 1H), 7.31 (dd, J=8.7, 7.0 Hz, 2H), 7.28~7.22 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 156.6, 148.5, 141.3, 140.8, 139.3, 135.6, 133.2, 132.8, 132.1, 129.2, 128.7, 128.6 (2C), 127.6 (2C), 125.9, 122.6; MS (ESI) m/z: 296.05 [M+H]+.
2-(2-(4-tert-Butyl-phenylsulfonyl)-phenyl)-pyridine
(3ad): Yield 78% (54.8 mg). Colorless viscous oil. 1H NMR (500 MHz, CDCl3) δ: 8.39~8.29 (m, 2H), 7.75~7.68 (m, 1H), 7.66~7.57 (m, 2H), 7.54 (d, J=7.8 Hz, 1H), 7.39~7.33 (m, 3H), 7.31~7.25 (m, 2H), 7.23 (dd, J=7.1, 5.2 Hz, 1H), 1.27 (s, 9H); 13C NMR (126 MHz, CDCl3) δ: 156.6, 156.5, 148.5, 140.6, 139.7, 138.1, 135.5, 133.1, 132.0, 128.9, 128.7, 127.5 (2C), 126.0, 125.7 (2C), 122.5, 35.1, 31.0 (3C). anal. calcd for C21H21NO2S: C 71.77, H 6.02, N 3.99; found C 71.64, H 6.07, N 4.05; MS (ESI) m/z: 352.23 [M+H]+.
2-(2-(4-Nitrophenylsulfonyl)-phenyl)-pyridine (3ae): Yield 18% (12.2 mg). Yellow solid, m.p. 121.4~123.6 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.38 (d, J=4.4 Hz, 1H), 8.03~7.96 (m, 2H), 7.64 (t, J=7.6 Hz, 1H), 7.58 (d, J=7.7 Hz, 1H), 7.53~7.48 (m, 5H), 7.46~7.40 (m, 1H), 7.22~7.15 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 154.0, 150.6, 149.6, 146.1, 140.1, 136.1, 133.6, 131.7, 130.3, 129.4 (2C), 128.3, 125.1, 124.1, 123.8 (2C), 122.3; MS (ESI): anal. calcd for C17H12N2O4S: C 59.99, H 3.55, N 8.23; found C 60.10, H 3.57, N 8.15; MS (ESI) m/z: 341.15 [M+H]+.
2-(2-(2-Nathphylsulfonyl)-phenyl)-pyridine (3af):[21] Yield 43% (29.7 mg). White solid, m.p. 111.6~114.9 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.41 (dd, J=6.3, 2.8 Hz, 1H), 8.15 (d, J=4.5 Hz, 1H), 7.92 (s, 1H), 7.80 (dd, J=14.4, 8.4 Hz, 2H), 7.76~7.68 (m, 2H), 7.66~7.56 (m, 3H), 7.56~7.48 (m, 3H), 7.35 (dd, J=5.9, 2.8 Hz, 1H), 7.17 (dd, J=7.0, 5.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ: 156.6, 148.3, 140.9, 139.30, 138.0, 135.5, 134.9, 133.2, 132.1, 131.8, 129.5, 129.3, 129.2, 129.1, 128.9, 128.8, 127.9, 127.3, 125.9, 122.7, 122.7; MS (ESI) m/z: 346.17 [M+H]+.
2-(2-(1-Nathphylsulfonyl)-phenyl)-pyridine (3ag): Yield 29% (20.0 mg). Viscous oil. 1H NMR (500 MHz, CDCl3) δ: 8.48 (d, J=7.7 Hz, 1H), 8.24~8.17 (m, 2H), 7.92 (d,
J=8.1 Hz, 1H), 7.82 (d, J=8.0 Hz, 1H), 7.69~7.57 (m, 4H), 7.50~7.40 (m, 3H), 7.30 (d, J=7.2 Hz, 1H), 7.22~7.12 (m, 2H); 13C NMR (126 MHz, CDCl3) δ: 156.1, 148.6, 140.4, 140.0, 135.3, 135.1, 134.6, 133.9, 133.0, 132.1, 129.9, 128.9, 128.7, 128.4, 128.3, 128.1, 126.7, 126.0, 124.0, 123.7, 122.5; anal. calcd for C21H15NO2S: C 73.02, H 4.38, N 4.06; found C 73.11, H 4.36, N 4.05; MS (ESI) m/z: 346.21 [M+H]+.
2-(2-(Thiophene-2-sulfony)-phenyl)-pyridine (3ah):[21] Yield 55% (33.1 mg). White solid, m.p. 100.4~102.4 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.53 (d, J=4.5 Hz, 1H), 8.29 (m, J=7.9, 1.2 Hz, 1H), 7.76 (td, J=7.7, 1.7 Hz, 1H), 7.65 (td, J=7.5, 1.3 Hz, 1H), 7.60 (dd, J=7.8, 1.4 Hz, 1H), 7.59~7.54 (m, 2H), 7.40 (dd, J=7.4, 1.3 Hz, 1H), 7.31 (m, J=7.5, 4.9, 0.9 Hz, 1H), 7.28 (dd, J=3.8, 1.3 Hz, 1H), 6.92 (dd, J=4.9, 3.9 Hz, 1H); 13CNMR (126 MHz, CDCl3) δ: 156.8, 148.5, 142.9, 140.7, 140.2, 135.8, 134.1, 133.8, 133.3, 132.1, 129.1, 128.9, 127.2, 125.7, 122.8; MS (ESI) m/z: 302.12 [M+H]+.
2-(2-(5-Chloro-thiophene-2-sulfony)-phenyl)-pyridine
(3ai): Yield 57% (38.2 mg). White solid, m.p. 133.3~136.2 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.60 (d, J=4.7 Hz, 1H), 8.26 (dd, J=8.0, 0.8 Hz, 1H), 7.79 (td, J=7.7, 1.7 Hz, 1H), 7.67 (td, J=7.5, 1.1 Hz, 1H), 7.60 (td, J=7.9, 1.2 Hz, 1H), 7.55 (d, J=7.8 Hz, 1H), 7.42 (dd, J=7.5, 1.0 Hz, 1H), 7.35 (dd, J=7.1, 5.3 Hz, 1H), 7.15 (d, J=4.1 Hz, 1H), 6.78 (d, J=4.1 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ: 156.9, 148.5, 140.9, 140.8, 139.8, 139.6, 136.0, 133.5(2C), 132.1, 129.4, 129.1, 126.6, 125.4, 122.9. anal. calcd for C15H10ClNO2S2: C 53.65, H 3.00, N 4.17; found C 53.70, H 3.03, N 4.08; MS (ESI) m/z: 336.05 [M+H]+.
2-(2-(4-tert-Butyl-phenylsulfonyl)-4-methyl-phenyl)-
pyridine (3bd): Yield 73% (53.3 mg). White solid. m.p. 175.0~177.0 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.32 (d, J=4.7 Hz, 1H), 8.19 (s, 1H), 7.71 (td, J=7.7, 1.7 Hz, 1H), 7.54 (d, J=7.8 Hz, 1H), 7.45 (d, J=7.7 Hz, 1H), 7.40~7.35 (m, 2H), 7.33~7.28 (m, 2H), 7.26 (d, J=7.7 Hz, 1H), 7.24~7.20 (m, 1H), 2.52 (s, 3H), 1.29 (s, 9H); 13C NMR (126 MHz, CDCl3) δ: 156.7, 156.3, 148.4, 139.3, 139.0, 138.2, 137.8, 135.5, 133.7, 132.0, 129.2, 127.5 (2C), 126.1, 125.6 (2C), 122.3, 35.1, 31.0 (3C), 21.3; anal. calcd for C22H23NO2S: C 72.30, H 6.34, N 3.83; found C 72.11, H 6.33, N 3.89; MS (ESI) m/z: 366.14 [M+H]+.
1-(2-(4-tert-Butylphenylsulfonyl)-phenyl)-1H-pyrazole
(3cd): Yield 28% (19.0 mg). Colorless viscous oil. 1H NMR (500 MHz, CDCl3) δ: 8.40 (dd, J=7.6, 1.8 Hz, 1H), 7.93 (d, J=2.4 Hz, 1H), 7.70~7.61 (m, 2H), 7.41 (d, J=1.5 Hz, 1H), 7.38~7.30 (m, 5H), 6.40 (t, J=2.1 Hz, 1H), 1.28 (s, 9H); 13C NMR (126 MHz, CDCl3) δ: 157.2, 140.8, 138.9, 137.6, 136.7, 134.4, 134.1, 130.3, 129.6, 129.2, 127.4 (2C), 125.8 (2C), 106.4, 35.1, 31.0 (3C). anal. calcd for C19H20N2O2S: C 67.03, H 5.92, N 8.23; found C 67.08, H 5.93, N 8.19; MS (ESI) m/z: 341.13 [M+H]+.
4.4 Catalyst recycle experiment
after each reaction cycle, the solvent, substrate, and products were removed by centrifugation; the separated catalyst was washed thoroughly with 0.1 mol/L NaOH ethanol solution (twice), distilled water (4 times), and then washed twice with ethanol followed by centrifugal separation and drying at 80 ℃ for 12 h. The recovered catalyst was used for the next cycle.
4.5 Hot filtration test
The hot filtration test was applied by filtering the model reaction mixture through a pre-heated celite pad after the reaction for 2 h. The filtrate was detected by GC to obtain the conversion of 1a and TOF. Then the filtered reaction solution continued to react for 2 h under normal conditions, and was detected by GC again.[18]
Supporting Information The 1H NMR and 13C NMR spectra of the products. The Supporting Information is available free of charge via the Internet athttp://sioc-journal.cn.
-
-
[1]
(a) Huang, Z.; Lim, H. N.; Mo, F.; Young, M. C.; Dong, G. Chem. Soc. Rev. 2015, 44, 7764.
(b) Segawa, Y.; Maekawa, T.; Itami, K. angew. Chem., Int. Ed. 2015, 54, 66.
(c) Ye, B.; Cramer, N. acc. Chem. Res. 2015, 48, 1308.
(d)(d) Daugulis, O.; Roane, J.; Tran, L. D. acc. Chem. Res. 2015, 48, 1053. -
[2]
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147. doi: 10.1021/cr900184e
-
[3]
(a) Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. angew. Chem., Int. Ed. 2009, 48, 5094.
(b)Daugulis, O.; Do, H.-Q.; Shabashov, D. acc. Chem. Res. 2009, 42, 1074.
(c)Ricci, P.; Krä mer, K.; Cambeiro, X. C.; Larrosa, I. J. am. Chem. Soc. 2013, 135, 13258.
(d)Sehnal, P.; Taylor, R. J. K.; Fairlamb, I. J. S. Chem. Rev. 2010, 110, 824. -
[4]
(a) Dick, a. R.; Hull, K. L.; Sanford, M. S. J. am. Chem. Soc. 2004, 126, 2300.
(b)Kalyani, D.; Deprez, N. R.; Desai, L. V.; Sanford, M. S. J. am. Chem. Soc. 2005, 127, 7330.
(c)Deprez, N. R.; Sanford, M. S. J. am. Chem. Soc. 2009, 131, 11234. -
[5]
Powers, D. C.; Ritter, T. Nat. Chem. 2009, 1, 302. doi: 10.1038/nchem.246
-
[6]
(a) Liu, Y.-J.; Xu, H.; Kong, W.-J.; Shang, M.; Dai, H.-X.; Yu, J.-Q. Nature 2014, 515, 389.
(b)Yu, L.; Huang, Y. P.; Wei, Z.; Ding, Y. H.; Su, C. L.; Xu, Q. J. Org. Chem. 2015, 80, 8677. -
[7]
(a) adak, L.; Bhadra, S.; Ranu, B. C. Tetrahedron Lett. 2010, 5, 3811.
(b)Williams, T. J.; Reay, a. J.; Whitwood, a. C.; Fairlamb, I. J. S. Chem. Commun. 2014, 50, 3052. -
[8]
(a) Basle, O.; Bidange, J.; Shuai, Q.; Li, C.-J. adv. Synth. Catal. 2010, 352, 1145.
(b)Zhao, X.; Dimitrijevic, E.; Dong, V. M. J. am. Chem. Soc. 2009, 131, 3466. -
[9]
(a) Trindade, a. F.; Gois, P. M. P.; afonso, C. a. M. Chem. Rev. 2009, 109, 418.
(b)Climent, M. J.; Corma, a.; Iborra, S. Chem. Rev. 2011, 111, 1072.
(c)Li, C. L.; Sato, T.; Yamauchi, Y. Chem. Commun. 2014, 50, 11753. -
[10]
(a) Sun, J. W.; Fu, Y. S.; He, G. Y.; Sun, X. Q.; Wang, X. Catal. Sci. Technol. 2014, 4, 1742.
(b) Mandegani, Z.; asadi, M.; asadi, Z.; Mohajeri, a.; Iranpoor, N.; Omidvar, a. Green Chem. 2015, 17, 3326. -
[11]
(a) Coupry, D. E.; Butson, J.; Petkov, P. S.; Saunders, M.; Donnell, K. O.; Kim, H.; Buckley, C.; addicoat, M.; Heine, T.; Szilágyi, P. Á . Chem. Commun. 2016, 52, 5175.
(b)Chen, L. Y.; Rangan, S.; Li, J.; Jianga, H. F.; Li, Y. W. Green Chem. 2014, 16, 3978.
(c)Fu, W. Q.; Feng, Y.; Fang, Z. X.; Chen, Q.; Tang, T.; Yua, Q. Y. Tang, T. D. Chem. Commun. 2016, 52, 3115. -
[12]
(a) Kishore, R. M.; Kantam, L.; Yadav, J.; Sudhakar, M.; Laha, S.; Venugopal, a. J. Mol. Catal. a: Chem. 2013, 379, 213.
(b)Jiao, Z. F.; Zhai, Z. Y.; Guo, X. N.; Guo, X. Y. J. Phys. Chem. C 2015, 119, 3238.
(c)Huang, J. P.; Wang, W.; Li, H. X. aCS Catal. 2013, 3, 1526. -
[13]
Zeng, M. F.; Du, Y. J.; Qi, C. Z.; Zuo, S. F.; Li, X. D.; Shao, L. J.; Zhang, X. M. Green Chem. 2011, 13, 350.
-
[14]
Hu, J. Y.; Yang, Q. W.; Yang, L. F.; Zhang, Z. G.; Su, B. G.; Bao, Z. B.; Ren, Q. L.; Xing, H. B.; Dai, S. aCS Catal. 2015, 5, 6724. doi: 10.1021/acscatal.5b01690
-
[15]
(a) aziz, J.; Messaoudi, S.; alami, M.; Hamze, a. Org. Biomol. Chem. 2014, 12, 9743.
(b)Xiao, F. H.; Chen, S. Q.; Tian, J. X.; Huang, H. W.; Liu, Y. J.; Deng, G. J. Green Chem. 2016, 18, 1538.
(c)Liang, S.; Zhang, R. Y.; Xi, L.Y.; Chen, S. Y.; Yu, X. Q. J. Org. Chem. 2013, 78, 11874.
(d)Reddy, L. R.; Hu, B.; Prashad, M.; Prasad, K. angew. Chem., Int. Ed. 2009, 48, 172.
(e)Taniguchi, N. J. Org. Chem. 2015, 80, 1764. -
[16]
Xu, Y. F.; Liu, P.; Li, S. L.; Sun. P. P. J. Org. Chem. 2015, 80, 1269.
-
[17]
Pillo, T.; Zimmermann, R.; Steiner, P.; Hüfner, S. J. Phys.: Condens. Matter 1997, 9, 3987. doi: 10.1088/0953-8984/9/19/018
-
[18]
Zhang, D.; Zhaorigetu, B.; Bao, Y. S. J. Phys. Chem. C 2015, 119, 20426. doi: 10.1021/acs.jpcc.5b04735
-
[19]
(a) Joo, S. H.; Park, J. Y.; Tsung, C. K.; Yamada, Y.; Yang, P. D. Nat. Mater. 2009, 8, 126.
(b) Qiao, Z. a.; Zhang, P. F.; Chai, S. H.; Chi, M. F.; Veith, G. M.; Gallego, N. C.; Kidder, M.; Dai, S. J. am. Chem. Soc. 2014, 136, 11260. -
[20]
Some report on the extensive Pd leaching of Pd/γ-al2O3, see:(a) Brazier, J. B.; Nguyen, B. N.; adrio, L. a.; Barreiro, E. M.; Leong, W. P.; Newton, M. a.; Figueroa, S. J. a.; Hellgardt, K.; Hii, K. K. M. Catal. Today 2014, 229, 95.
(b)Thathagar, M. B.; ten Elshof, J. E.; Rothenberg, G. angew. Chem., Int. Ed. 2006, 45, 2886. -
[21]
Wang, F.; Yu, X.; Qi, Z.; Li, X. Chem.-Eur. J. 2016, 22, 511. doi: 10.1002/chem.201504179
-
[22]
Niu, L.; Yang, H.; Yang, D.; Fu, H. adv. Synth. Catal. 2012, 354, 2211.
-
[1]
-
Figure 1 XPS spectra of fresh and used Pd/γ-al2O3 (wPd=3%) (after reaction different times)
Figure 2 Photos of fresh and used Pd/γ-al2O3 (wPd=3%) (after reaction different times)
Figure 4 TEM images of fresh (a), after 1th cycle (b) and after 3th cycle (c) Pd/γ-al2O3 (wPd=3%), and the PdNPs size distributions of fresh (error range: ±0.25 nm) (d), after 1th cycle (Error range: ±1.0 nm) (e) and after 3th cycle (Error range: ±1.0 nm) (f) catalysts, respectively
Table 1. Optimization of reaction conditionsa
Entry Catalyst Temp./℃ Yield/% 1 Pd/γ-al2O3 (wPd=3%) 125 73 2 Pd(Oac)2 125 75 3 Pd/NiO (wPd=3%) 125 33 4 Pd/TiO2 (wPd=3%) 125 52 5 Pd/ZrO2 (wPd=3%) 125 56 6 Pd/γ-al2O3 (wPd=1%) 125 54 7 Pd/γ-al2O3 (wPd=5%) 125 61 8 γ-al2O3 125 0 10 Pd/γ-al2O3 (wPd=3%) 130 70 11 Pd/γ-al2O3 (wPd=3%) 115 65 a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), catalyst (35 mg), K2CO3 (0.4 mmol), and ethylbenzene (2 mL) at 125 ℃ for 24 h. b Isolated yield.  下载: 导出CSV
下载: 导出CSV
Table 2. Substrate scope of ortho-directed sulfonylationa
Entry 1 2 Product Yieldb/% Entry 1 2 Product Yieldb/% 1 1a 51 6 1a 29 2 1a 48 7 1a 55 3 1a 78 8 1a 57 4 1a 18 9 1b 73 5 1a 43 10 1c 28 a Reaction conditions: 1 (0.2 mmol), 2 (0.6 mmol), Pd/γ-al2O3 (wPd=3%, 35 mg), K2CO3 (0.4 mmol), ethylbenzene (2 mL), 125 ℃, 24 h. b Isolated yield.
下载: 导出CSV
Table 3. Characterization results of aaS of different Pd/γ-al2O3 catalysts
Sample Pd loading w/% Pd leaching
rate/%3% Pd/γ-al2O3 (fresh) 3.01 3% Pd/γ-al2O3 (after 1th cycle) 2.36 -22 3% Pd/γ-al2O3 (after 3th cycle) 0.51 -83
下载: 导出CSV
Table 4. The results of the hot filtration test
Time/h Conversiona/% TOF 0 0 2 27.9 2.81 4 (after hot filtration) 68.0 6.86
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 9
- 文章访问数: 1212
- HTML全文浏览量: 130
