Table 1.
Screening reaction conditionsa
Citation:
Yu Jiao, Lin Jinhong, Xiao Jichang. ZnO-Promoted Wittig gem-Difluoroolefination of Aldehydes with [Ph3P+CF2H·Br-][J]. Chinese Journal of Organic Chemistry,
2019, 39(1): 265-269.
doi:
10.6023/cjoc201806024

氧化锌促进的醛与[Ph3P+CF2H·Br-]的Wittig偕二氟烯基化反应
English
ZnO-Promoted Wittig gem-Difluoroolefination of Aldehydes with [Ph3P+CF2H·Br-]
Abstract:
Wittig gem-difluoroolefination of aldehydes with difluoromethyl phosphonium salt (Ph3P+CF2H·Br-) by using zinc oxide as a base is described. Although the proton in the CF2H group is acidic and a base could easily lead to its deprotonation to form ylide (Ph3P+CF2-), the attack of the base at the positive phosphorus atom may also take place to produce a nucleophilic [HCF2-] equivalent, and then nucleophilic difluoromethylation instead of Wittig reaction would occur. The use of ZnO as the base favored the Wittig reaction and the nucleophilic difluoromethylation was not observed. Furthermore, the excessive ZnO and ZnⅡ salts produced from ZnO could be easily removed by filtration, which may be convenient for the purification process.
-
Key words:
- Wittig
- / gem-difluoroolefination
- / aldehydes
- / difluoromethyl phosphonium salt
- / zinc oxide
-
1. Introduction
Due to the unique properties of fluorine element such as strong electronegativity and small atomic radius, the incorporation of fluorine atom(s) or fluorine-containing group into organic molecules, especially pharmaceuticals, may result in profound modification of their physicochemical properties, including lipophilicity, membrane permeability, and binding selectivity to target receptors in the body.[1] gem-Difluorovinyl group is a valuable fluorine-containing moiety and gem-difluoroolefin derivatives have been considered as potential enzyme inhibitors.[2] Besides, the gem-difluorovinyl group is very electrophilic and could be conveniently transformed further to afford other useful products.[3] Consequently, significant efforts have been devoted to the development of efficient methods for the synthesis of gem-difluoroolefins.[4] Many strategies have been well established, such as β-elimination of functionalized difluoromethyl compounds, [5] transition metal-catalyzed coupling with gem-difluorovinyl-containing reagents, [3a, 4c, 6] and gem-difluoroolefination of carbonyl compounds including Julia reaction, [7] Horner-Wadsworth-Emmons reaction[8] and Wittig reaction.[9] Obviously, Wittig reaction is one of the most attractive approaches due to its high efficiency and mild reaction conditions.
Phsphonium ylide (R3P+CF2-) is a key intermediate in the Wittig gem-difluoroolefination reaction. It can be generated from difluoromethylene phosphonium salts such as [PH3P+CF2CO2-], [10] [Ph3P+CF2H•X-][11] or [R3P+CF2-Br•X-], [12] or generated by a reaction of difluorocarbene with a trivalent phosphine.[9b, 9c, 13] PH3P+CF2CO2-, an efficient phosphonium ylide reagent developed by us recently, [10b, 13, 14] could readily undergo decarboxylation under warming conditions to generate the phosphonium ylide. We found that decarboxylation of PH3P+CF2CO2- under acidic conditions could afford difluoromethyl phosphonium salts [Ph3P+CF2H•X-].[15] It is reasonable to conceive that phosphonium ylide can be produced from this difluoromethyl salt via deprotonation by a base, and Wittig reaction of aldehydes would then be easily achieved. Interestingly, our investigation revealed that aldehydes may react with the phosphonium salt by a Wittig reaction process[11] or a nucleophilic difluoromethylation process, [15] depending on the base used. A base does not necessarily deprotonate the phosphonium salt to form ylide. The attack of the base at the phosphorus atom may occur to generate a nucleophilic [HCF2-] equivalent and thus the difluoromethylation instead of Wittig reaction would take place.[15] 1, 8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was found to be a suitable base for the Wittig reaction path.[11] But the use of this organic base may result in an inconvenient purification process since a tedious procedure such as flash column chromatography or extraction may be needed to remove excessive DBU and its protonated form. We found that zinc oxide (ZnO) could act as an efficient base to achieve Wittig gem-difluoroolefination of aldehydes. Nucleophilic difluoromethylation was suppressed and both excessive ZnO and ZnⅡ salts produced from ZnO could be easily removed by filtration. The preliminary results are described herein.
2. Results and discussion
Various metal oxides were screened in our initial attempts at the Wittig gem-difluoroolefination of aldehyde 1a with phosphonium salt 2 (Table 1, Entries 1~6) and it was found that ZnO could well promote the reaction (Entry 6). Unlike ZnO, other zinc salts such as ZnCl2 and Zn(OH)2 were not effective (Entries 7~9). A brief survey of the reaction solvent (Entries 10~13) revealed that a good yield could be obtained in a polar solvent such as N, N-dimethylformamide (DMF) or N, N-dimethylacetamide (DMAc) (Entries 6 and 7). The reaction temperature has an important effect. Lowering the reaction temperature to 80 ℃ dramatically suppressed the desired process (Entry 14). A comparable yield was obtained at 90 ℃ or 100 ℃ (Entries 15~16), and further elevating to 140 ℃ led to the decrease in the yield (Entry 17). ZnO has to be used in the same equivalent with that of salt 2. Otherwise the yield would be decreased (Entry 18 vs. Entry 6). Three equivalent of phosphonium salt 2 was necessary. Low yields would be obtained by decreasing the loading of this salt (Entries 19~20). To our delight, the presence of 4Å molecular sieves gave the desired product in 91% yield (Entry 21). No reaction was observed without the presence of ZnO, reflecting the important role of ZnO (Entry 22).
Table 1

 下载:
导出CSV
下载:
导出CSV
Entry Base 1a:2:
basebSolvent Temp./℃ Yieldc/% 1 NaO 1:3:3 DMF 120 13 2 MgO 1:3:3 DMF 120 ND 3 Al2O3 1:3:3 DMF 120 ND 4 CaO 1:3:3 DMF 120 ND 5 CuO 1:3:3 DMF 120 ND 6 ZnO 1:3:3 DMF 120 70 7 ZnCl2 1:3:3 DMF 120 ND 8 Zn(OH)2 1:3:3 DMF 120 26 9 ZnF2 1:3:3 DMF 120 10 10 ZnO 1:3:3 DMAc 120 70 11 ZnO 1:3:3 CH3CN 120 68 12 ZnO 1:3:3 Tetrahy-drofuran 120 38 13 ZnO 1:3:3 Toluene 120 19 14 ZnO 1:3:3 DMF 80 36 15 ZnO 1:3:3 DMF 90 74 16 ZnO 1:3:3 DMF 100 74 17 ZnO 1:3:3 DMF 140 63 18 ZnO 1:3:1 DMF 120 54 19 ZnO 1:1:1 DMF 120 21 20 ZnO 1:2:2 DMF 120 46 21d ZnO 1:3:3 DMF 90 91 22 — 1:3:0 DMF 90 ND a Reaction conditions: 1a (0.2 mmol), salt 2, base and solvent (1.5 mL) at the indicated temperature for 10 h; b Molar ratio of 1a:2:base; c The yields were determined by 19F NMR spectroscopy; d 15 h of reaction time was used, and 300 mg 4 Å molecular sieves were added. With the optimized reaction conditions in hand (Table 1, Entry 18), then the substrate scope of the Wittig gem-difluoroolefination of aldehydes was investigated. As shown in Table 2, electron-deficient and electron-neutral aryl aldehydes could all be converted smoothly into de- sired products in good yields (3a~3e, 3h~3k). In the case of electron-rich aryl aldehydes, moderate yields were obtained (3f~3g), probably due to the deactivation of the carbonyl group. Various functional groups were tolerated, such as halide, nitro, cyanide and terminal vinyl groups. Compared with aryl aldehydes, alkyl aldehydes showed lower reactivity and the expected product was obtained in a low yield (3l). α, β-unsaturated aldehydes could also be well transformed into the final products in moderate yields (3m~3p).
Table 2
Since we have previously found that aldehyde may react with phosphonium salt 2 to afford difluoromethyl alco-hols, [15] it is reasonable to question that if olefins are gen- erated from difluoromethyl alcohols. It was found that difluoromethyl alcohol 4a remained intact in the presence of ZnO, indicating that the path via alcohols could be excluded (Eq. 1). Therefore, the reaction should proceed via phosphonium ylide PH3P+CF2- formed by deprotonation of phosphonium salt 2 with ZnO. Indeed, salt 2 could react with ZnO and many unknown fluoro-species were detected by 19F NMR spectroscorpy (Eq. 2). The reason for generating unknown species is that ylide PH3P+CF2- is highly active and would readily undergo side reactions without the presence of a substrate.

(1) 
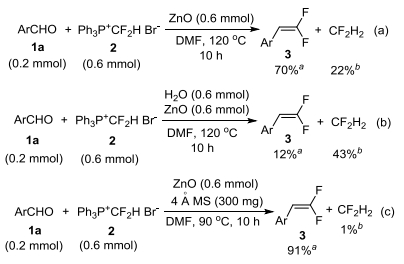
(2) Since difluoromethyl phosphonium salt 2 was deprotonated by ZnO, the question arises as to whether the hydroxide in Zn(OH)2 or ZnBr(OH) produced from ZnO was also a base for deprotonation. We believe that the hydroxide does not act as a base taking into account the following experimental evidence. Firstly, if it is a base, ZnO would not be needed to be used in stoichiometric amount compared with salt 2. But a decreased loading of ZnO led to the decrease in the yield (Table 1, Entry 15 vs. Entry 6). Secondly, our previous observation of nucleophilic reaction with phosphonium salts indicates that hydroxide anion may prefer to attack the positive phosphonium atom.[15, 16] This process would generate a nucleophilic [HCF2-] equivalent, which may be quenched by water to produce H2CF2. Indeed, although 70% yield could be obtained in the absence of 4Å molecular sieves, a large amount of H2CF2 was also formed (Scheme 1a), which may be because trace amount of water is present in the reaction system. The external addition of water almost completely suppressed the Wittig reaction, and H2CF2 was produced as a major byproduct (Scheme 1b). Water might be favorable for dissolving zinc salt [Zn(OH)2 and ZnBr(OH)], and then the attack of hydroxide at positive phosphonium turned out to be a dominate path. Almost no H2CF2 was observed in the presence of 4Å molecular sieves (Scheme 1c), further supporting that hydroxide would not protonate phosphonium salt 2.
Scheme 1
 Scheme 1. Effects of water
Scheme 1. Effects of watera The yield was determined by 19F NMR spectroscopy based on aldehyde 1a as the limiting reagent; b the yield was determined by 19F NMR spectroscopy based on salt 2 as the limiting reagent.
3. Conclusions
In summary, we have developed ZnO-promoted Wittig gem-difluoroolefination of aldehydes with phosphonium salt [Ph3P+CF2H•Br-]. Although the phosphonium salt may act as a [HCF2-] equivalent to attack aldehyde, this nucleophilic difluoromethylation process was suppressed by using ZnO as the base. The excessive ZnO and ZnⅡ salts produced from ZnO could be easily removed by filtration, which may be convenient for the purification process.
4. Experimental section
4.1 General information
1H NMR, 13C NMR and 19F NMR spectra were detected on a 500, 400 or 300 MHz NMR spectrometer. Mass spectra were obtained on GC-MS or LC-MS (ESI). High resolution mass data were recorded on a high resolution mass spectrometer in the EI or ESI mode. The mass analyzer types for HRMS-EI, HRMS-ESI, and HRMSMALDI are time-of-flight, Fourier transform mass spectrometer, and Fourier transform mass spectrometer, respectively. Unless otherwise noted, all reagents were obtained commercially and used without further purification.
4.2 Typical procedure for the reaction of phosphonium salts with aldehyde (products 3a, 3b, 3c, 3e, 3h, 3i, 3l, 3m, 3n, 3o and 3p)
Into a 20 mL sealed tube were added aldehyde 1a (1.0 mmol, 182.2 mg, 1.0 equiv.), phosphonium salts 2 (3 mmol, 1179.6 mg, 3 equiv.), ZnO (3 mmol, 244.2 mg, 3 equiv.), 4 Å molecular sieves (1.5 g) and DMF (7.5 mL) under a N2 atmosphere. The tube was sealed and the reaction mixture was stirred at 90 ℃ for 10 h. After being cooled to room temperature, the mixture was directly subjected to flash column chromatography to afford the final product.
4-(2, 2-Difluorovinyl)-1, 1'-biphenyl (3a):[11] 90% yield. 1H NMR (400 MHz, CDCl3) δ: 7.63~7.55 (m, 4H), 7.48~7.32 (m, 5H), 5.32 (dd, J=26.3, 3.8 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -81.87 (dd, J=30.6, 26.3 Hz, 1F), -83.81 (dd, J=30.6, 3.8 Hz, 1F).
1-Bromo-4-(2, 2-difluorovinyl)benzene (3b):[11] 81% yield. 1H NMR (400 MHz, CDCl3) δ: 7.45 (d, J=8.5, 2H), 7.19 (d, J=8.5, 2H), 5.22 (dd, J=25.9, 3.6 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -81.28 (dd, J=29.2, 25.9 Hz, 1F), -83.12 (dd, J=29.2, 3.6 Hz, 1F).
1-(2, 2-Difluorovinyl)-4-iodobenzene (3c):[17] 90% yield. 1H NMR (400 MHz, CDCl3) δ: 7.65 (d, J=8.3 Hz, 2H), 7.06 (d, J=8.3 Hz, 2H), 5.21 (dd, J=26.5, 3.5 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -80.83 (t, J=26.5, 1F), -82.75 (dd, J=26.5, 3.5 Hz, 1F).
4-(2, 2-Difluorovinyl)benzonitrile (3e):[11] 62% yield. 1H NMR (400 MHz, CDCl3) δ: 7.63 (d, J=8.4 Hz, 2H), 7.43 (d, J=8.4 Hz, 2H), 5.34 (dd, J=25.6, 3.4 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -77.80 (dd, J=25.6, 20.4 Hz, 1F), -79.47 (dd, J=20.4, 3.4 Hz, 1F).
2-(2, 2-Difluorovinyl)naphthalene (3h)[11]: 80% yield. 1H NMR (400 MHz, CDCl3) δ: 7.83~7.72 (m, 4H), 7.51~7.41 (m, 3H), 5.43 (dd, J=26.2, 3.8 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -81.95 (dd, J=30.8, 26.2 Hz, 1F), -83.67 (dd, J=30.8, 3.8 Hz, 1F).
9-(2, 2-Difluorovinyl)anthracene (3i): 72% yield. Yellow solid, m.p. 100.2~101.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.38 (s, 1H), 8.09 (d, J=8.7 Hz, 2H), 7.95 (d, J=7.8 Hz, 2H), 7.57~7.38 (m, 4H), 5.92 (d, J=26.4 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -81.72 (t, J=26.4 Hz, 1F), -84.31 (d, J=26.4 Hz, 1F); 13C NMR (101 MHz, CDCl3) δ: 155.79 (dd, J=294.0, 290.6 Hz), 131.41 (s), 130.35 (s), 128.89 (s), 127.73 (s), 126.16 (s), 125.39 (s), 125.30 (s), 122.54 (dd, J=6.9, 2.3 Hz), 76.43 (dd, J=28.5, 20.1 Hz); IR (neat) ν: 1732, 1316, 1302, 1193, 1179, 1070, 917, 931, 890, 872, 804, 788, 740, 727, 639, 528 cm-1; HRMS (EI) Calcd for C16H10F2 [M]+ 240.0751; found 240.0756.
(4, 4-Difluorobut-3-en-1-yl)benzene (3l)[10b]: 34% yield. 1H NMR (400 MHz, CDCl3) δ: 7.33~7.14 (m, 5H), 4.15 (dt, J=25.4, 7.8 Hz, 1H), 2.68 (t, J=7.6 Hz, 2H), 2.34~2.22 (m, 2H); 19F NMR (376 MHz, CDCl3) δ: -89.04 (d, J=47.3 Hz, 1F), -91.08 (dd, J=47.3, 25.4 Hz, 1F).
(E)-1-Bromo-2-(4, 4-difluorobuta-1, 3-dien-1-yl)benzene (3m): 57% yield. 1H NMR (400 MHz, CDCl3) δ: 7.59~7.43 (m, 2H), 7.31~7.20 (m, 1H), 7.12~7.03 (m, 1H), 6.83 (d, J=15.8 Hz, 1H), 6.60 (dd, J=15.8, 10.9 Hz, 1H), 5.20 (dddd, J=23.7, 10.9, 1.4, 0.6 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -84.35 (t, J=23.7 Hz, 1F), -85.82 (d, J=23.7 Hz, 1F); 13C NMR (101 MHz, CDCl3) δ: 157.13 (dd, J=298.0, 292.6 Hz), 136.64 (s), 133.09 (s), 129.64 (dd, J=11.7, 3.4 Hz), 128.84 (s), 127.54 (s), 126.49 (s), 123.58 (s), 120.56 (dd, J=4.7, 2.3 Hz), 82.94 (dd, J=27.9, 16.7 Hz); IR (neat) ν: 2925, 1867, 1716, 1625, 1467, 1437, 1352, 1296, 1264, 1185, 1113, 1043, 1024, 961, 934, 847, 749 cm-1; HRMS (EI) Calcd for C10H7BrF2 243.9699; found 243.9697.
(E)-(4, 4-Difluorobuta-1, 3-dien-1-yl)benzene (3n)[11]: 68% yield. 1H NMR (400 MHz, CDCl3) δ: 7.42~7.18 (m, 5H), 6.66 (dd, J=15.9, 10.9 Hz, 1H), 6.47 (d, J=15.9 Hz, 1H), 5.14 (dd, J=26.5, 10.9 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -85.32 (t, J=26.5 Hz, 1F), -87.03 (d, J=26.5 Hz, 1F).
(E)-1-Bromo-4-(4, 4-difluorobuta-1, 3-dien-1-yl)benzene (3o)[18]: 68% yield.1H NMR (500 MHz, CDCl3) δ: 7.43 (d, J=8.4 Hz, 2H), 7.24 (dd, J=8.4 Hz, 2H), 6.64 (dd, J=15.9, 10.9 Hz, 1H), 6.40 (d, J=15.9 Hz, 1H), 5.12 (dd, J=23.9, 10.9 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -84.59 (t, J=23.9 Hz, 1F), -86.20 (d, J=23.9 Hz, 1F).
(E)-1-(4, 4-Difluorobuta-1, 3-dien-1-yl)-4-methylbenzene (3p)[18]: 38% yield. 1H NMR (400 MHz, CDCl3) δ: 7.27 (d, J=8.1 Hz, 2H), 7.12 (d, J=8.1 Hz, 2H), 6.60 (dd, J=15.9, 10.8 Hz, 1H), 6.44 (d, J=15.9 Hz, 1H), 5.11 (dd, J=24.3, 10.8 Hz, 1H), 2.33 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -85.82 (dd, J=27.8, 24.3 Hz, 1F), -87.61 (d, J=27.8 Hz, 1F).
4.3 Typical procedure for the reaction of phosphonium salt with aldehydes (products 3d, 3f, 3g, 3j and 3k)
Into a 20 mL sealed tube were added aldehyde 1d (1.0 mmol, 151.0 mg, 1.0 equiv.), phosphonium salt 2 (3 mmol, 1179.6 mg, 3 equiv.), ZnO (3 mmol, 244.2 mg, 3 equiv.), 4 Å molecular sieves (1.5 g) and DMF (7.5 mL) under a N2 atmosphere. The tube was sealed and the reaction mixture was stirred at 90 ℃ for 10 h. The solid was removed by filtration. The filtrate was diluted with water (50 mL) and then the crude product was extracted with CH2Cl2 (20 mL×3). The combined organic layers were washed with water (20 mL×3). The organic layer was dried over Na2SO4. The solvent was removed by concentration under reduced pressure, and the residue was subjected to flash column chromatography to afford the final product.
1-(2, 2-Difluorovinyl)-3-nitrobenzene (3d):[17] 75% yield. 1H NMR (400 MHz, CDCl3) δ: 8.19 (s, 1H), 8.10 (d, J=8.0 Hz, 1H), 7.65 (d, J=8.0 Hz, 1H), 7.53 (t, J=8.0 Hz, 1H), 5.39 (dd, J=24.5, 3.1 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -78.99 (t, J=24.5 Hz, 1F), -80.73 (dd, J=24.5, 3.1 Hz, 1F).
1-(2, 2-Difluorovinyl)-4-methoxybenzene (3f):[11] 56% yield. 1H NMR (400 MHz, CDCl3) δ: 7.25 (d, J=9.0 Hz, 2H), 6.87 (d, J=8.8 Hz, 2H), 5.21 (dd, J=26.4, 3.9 Hz, 1H), 3.80 (s, 3H); 19F NMR (376 MHz, CDCl3) δ: -84.69 (dd, J=36.8, 26.4 Hz, 1F), -86.50 (dd, J=36.8, 3.9 Hz, 1F).
1-(Allyloxy)-4-(2, 2-difluorovinyl)benzene (3g):[10b] 49% yield. 1H NMR (400 MHz, CDCl3) δ: 7.24 (d, J=8.9 Hz, 2H), 6.88 (d, J=8.9 Hz, 2H), 6.10~5.99 (m, 1H), 5.41 (d, J=17.2 Hz, 1H), 5.29 (d, J=10.5 Hz, 1H), 5.20 (dd, J=26.4, 3.8 Hz, 1H), 4.53 (dt, J=5.3, 1.5 Hz, 2H); 19F NMR (376 MHz, CDCl3) δ: -84.55 (dd, J=36.5, 26.4 Hz, 1F), -86.38 (dd, J=36.5, 3.8 Hz, 1F).
3-(2, 2-Difluorovinyl)benzo[b]thiophene (3j):[11] 70% yield. 1H NMR (400 MHz, CDCl3) δ: 7.86 (d, J=7.4 Hz, 1H), 7.70 (d, J=7.4 Hz, 1H), 7.45 (s, 1H), 7.44~7.34 (m, 2H), 5.59 (dd, J=25.7, 2.2 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ: -80.70 (t, J=25.7 Hz, 1F), -84.54 (dd, J=25.7, 2.2 Hz, 1F).
2-(2, 2-Difluorovinyl)benzofuran (3k)[10b]: 64% yield. 1H NMR (400 MHz, CDCl3) δ: 7.52 (d, J=7.2 Hz, 1H), 7.43 (d, J=8.1 Hz, 1H), 7.28~7.17 (m, 2H), 6.66 (s, 1H), 5.44 (dd, J=25.1, 1.8 Hz, 1H); 19F NMR (376 MHz, CDCl3) δ:-75.95 (dd, J=25.1, 16.2 Hz, 1F), -82.21 (dd, J=16.2, 1.8 Hz, 1F).
Supporting Information: 1H/19F/13C NMR spectra of products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
-
-
[1]
(a) Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology, Blackwell Publishing, Chichester, 2009.
(b) Meanwell, N. A. J. Med. Chem. 2011, 54, 2529.
(c) Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432.
(d) Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422. -
[2]
(a) McDonald, I. A.; Lacoste, J. M.; Bey, P.; Palfreyman, M. G.; Zreika, M. J. Med. Chem. 1985, 28, 186.
(b) Weintraub, P. M.; Holland, A. K.; Gates, C. A.; Moore, W. R.; Resvick, R. J.; Bey, P.; Peet, N. P. Bioorg. Med. Chem. 2003, 11, 427.
(c) Altenburger, J.-M.; Lassalle, G. Y.; Matrougui, M.; Galtier, D.; Jetha, J.-C.; Bocskei, Z.; Berry, C. N.; Lunven, C.; Lorrain, J.; Herault, J.-P.; Schaeffer, P.; O'Connor, S. E.; Herbert, J.-M. Bioorg. Med. Chem. 2004, 12, 1713. -
[3]
(a) Nguyen, B. V.; Burton, D. J. J. Org. Chem. 1997, 62, 7758.
(b) Yokota, M.; Fujita, D.; Ichikawa, J. Org. Lett. 2007, 9, 4639.
(c) Qiao, Y.; Si, T.; Yang, M.-H.; Altman, R. A. J. Org. Chem. 2014, 79, 7122. -
[4]
(a) Tozer, M. J.; Herpin, T. F. Tetrahedron 1996, 52, 8619.
(b) Burton, D.; Yang, Z.-Y.; Qiu, W. Chem. Rev. 1996, 96, 1641.
(c) Ichikawa, J. J. Fluorine Chem. 2000, 105, 257.
(d) Chelucci, G. Chem. Rev. 2012, 112, 1344. -
[5]
(a) Mae, M.; Amii, H.; Uneyama, K. Tetrahedron Lett. 2000, 41, 7893.
(b) Amii, H.; Kobayashi, T.; Terasawa, H.; Uneyama, K. Org. Lett. 2001, 3, 3103.
(c) Ichikawa, J.; Ishibashi, Y.; Fukui, H. Tetrahedron Lett. 2003, 44, 707.
(d) Ichikawa, J.; Fukui, H.; Ishibashi, Y. J. Org. Chem. 2003, 68, 7800.
(e) Miura, T.; Ito, Y.; Murakami, M. Chem. Lett. 2008, 37, 1006. -
[6]
(a) Crowley, P. J.; Howarth, J. A.; Owton, W. M.; Percy, J. M.; Stansfield, K. Tetrahedron Lett. 1996, 37, 5975.
(b) Goegsig, T. M.; Soebjerg, L. S.; Lindhardt, A. T.; Jensen, K. L.; Skrydstrup, T. J. Org. Chem. 2008, 73, 3404. -
[7]
(a) Prakash, G. K. S.; Wang, Y.; Hu, J.; Olah, G. A. J. Fluorine Chem. 2005, 126, 1361.
(b) Zhao, Y.; Huang, W.; Zhu, L.; Hu, J. Org. Lett. 2010, 12, 1444.
(c) Wang, X.-P.; Lin, J.-H.; Xiao, J.-C.; Zheng, X. Eur. J. Org. Chem. 2014, 928.
(d) Cao, C.-R.; Ou, S.; Jiang, M.; Liu, J.-T. Tetrahedron Lett. 2017, 58, 482. -
[8]
(a) Edwards, M. L.; Stemerick, D. M.; Jarvi, E. T.; Matthews, D. P.; McCarthy, J. R. Tetrahedron Lett. 1990, 31, 5571.
(b) Piettre, S. R.; Cabanas, L. Tetrahedron Lett. 1996, 37, 5881. -
[9]
(a) Serafinowski, P. J.; Brown, C. A. Tetrahedron 2000, 56, 333.
(b) Nowak, I.; Robins, M. Org. Lett. 2005, 7, 721.
(c) Thomoson, C. S.; Martinez, H.; Dolbier, W. R., Jr. J. Fluorine Chem. 2013, 150, 53.
(d) Wang, F.; Li, L.; Ni, C.; Hu, J. Beilstein J. Org. Chem. 2014, 10, 344. -
[10]
(a) Herkes, F.; Burton, D. J. Org. Chem. 1967, 1311.
(b) Zheng, J.; Cai, J.; Lin, J.-H.; Guo, Y.; Xiao, J.-C. Chem. Commun. 2013, 49, 7513. -
[11]
Li, Q.; Lin, J.-H.; Deng, Z.-Y.; Zheng, J.; Cai, J.; Xiao, J.-C. J. Fluorine Chem. 2014, 163, 38. doi: 10.1016/j.jfluchem.2014.04.011
-
[12]
(a) Naae, D. G.; Burton, D. J. J. Fluorine Chem. 1971, 1, 123.
(b) Naae, D. G.; Burton, D. J. Synth. Commun. 1973, 3, 197. -
[13]
Zheng, J.; Lin, J.-H.; Cai, J.; Xiao, J.-C. Chem.-Eur. J. 2013, 19, 15261. doi: 10.1002/chem.201303248
-
[14]
(a) Deng, X.-Y.; Lin, J.-H.; Zheng, J.; Xiao, J.-C. Chem. Commun. 2015, 51, 8805.
(b) Zheng, J.; Lin, J.-H.; Deng, X.-Y.; Xiao, J.-C. Org. Lett. 2015, 17, 532.
(c) Zheng, J.; Lin, J.-H.; Yu, L.-Y.; Wei, Y.; Zheng, X.; Xiao, J.-C. Org. Lett. 2015, 17, 6150.
(d) Zheng, J.; Wang, L.; Lin, J.-H.; Xiao, J.-C.; Liang, S. H. Angew. Chem., Int. Ed. 2015, 54, 13236.
(e) Zheng, J.; Cheng, R.; Lin, J.-H.; Yu, D. H.; Ma, L.; Jia, L.; Zhang, L.; Wang, L.; Xiao, J.-C.; Liang, S. H. Angew. Chem., Int. Ed. 2017, 56, 3196.
(f) Yu, J.; Lin, J.-H.; Xiao, J.-C. Angew. Chem., Int. Ed. 2017, 56, 16669. -
[15]
Deng, Z.; Lin, J.-H.; Cai, J.; Xiao, J.-C. Org. Lett. 2016, 18, 3206. doi: 10.1021/acs.orglett.6b01425
-
[16]
(a) Deng, Z.; Lin, J.-H.; Xiao, J.-C. Nat. Commun. 2016, 7, 10337.
(b) Deng, Z.; Liu, C.; Zeng, X.-L.; Lin, J.-H.; Xiao, J.-C. J. Org. Chem. 2016, 81, 12084. -
[17]
Nenajdenko, V. G.; Varseev, G. N.; Korotchenko, V. N.; Shastin, A. V.; Balenkova, E. S. J. Fluorine Chem. 2003, 124, 115-118. doi: 10.1016/S0022-1139(03)00199-4
-
[18]
Ichitsuka, T.; Takanohashi, T.; Fujita, T.; Ichikawa, J. J. Fluorine Chem. 2015, 170, 29. doi: 10.1016/j.jfluchem.2014.12.003
-
[1]
-
Scheme 1 Effects of water
a The yield was determined by 19F NMR spectroscopy based on aldehyde 1a as the limiting reagent; b the yield was determined by 19F NMR spectroscopy based on salt 2 as the limiting reagent.
Table 1. Screening reaction conditionsa
Entry Base 1a:2:
basebSolvent Temp./℃ Yieldc/% 1 NaO 1:3:3 DMF 120 13 2 MgO 1:3:3 DMF 120 ND 3 Al2O3 1:3:3 DMF 120 ND 4 CaO 1:3:3 DMF 120 ND 5 CuO 1:3:3 DMF 120 ND 6 ZnO 1:3:3 DMF 120 70 7 ZnCl2 1:3:3 DMF 120 ND 8 Zn(OH)2 1:3:3 DMF 120 26 9 ZnF2 1:3:3 DMF 120 10 10 ZnO 1:3:3 DMAc 120 70 11 ZnO 1:3:3 CH3CN 120 68 12 ZnO 1:3:3 Tetrahy-drofuran 120 38 13 ZnO 1:3:3 Toluene 120 19 14 ZnO 1:3:3 DMF 80 36 15 ZnO 1:3:3 DMF 90 74 16 ZnO 1:3:3 DMF 100 74 17 ZnO 1:3:3 DMF 140 63 18 ZnO 1:3:1 DMF 120 54 19 ZnO 1:1:1 DMF 120 21 20 ZnO 1:2:2 DMF 120 46 21d ZnO 1:3:3 DMF 90 91 22 — 1:3:0 DMF 90 ND a Reaction conditions: 1a (0.2 mmol), salt 2, base and solvent (1.5 mL) at the indicated temperature for 10 h; b Molar ratio of 1a:2:base; c The yields were determined by 19F NMR spectroscopy; d 15 h of reaction time was used, and 300 mg 4 Å molecular sieves were added.  下载: 导出CSV
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 14
- 文章访问数: 1647
- HTML全文浏览量: 172
