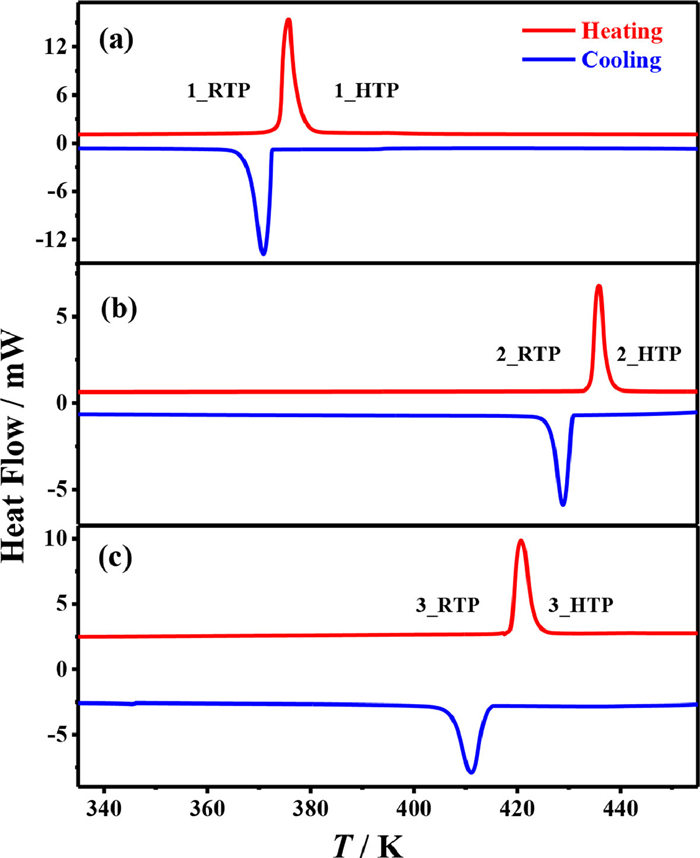
Figure 1.
DSC curves of (a) 1, (b) 2 and (c) 3 in a heating-cooling cycle.
Ferroelastic phase transitions in three new layered perovskites: (3-XC6H5CH2CH2NH3)2[CdCl4] (X = F, Cl, and Br)
Yaping Gong , Xiaoxian Chen , Bingqing Zhao , Jun Wang , Weixiong Zhang , Xiaoming Chen

Ferroelastic transitions are featured in spontaneous strain as order parameter [1–3]. Different orientations of ferroelastic domain, a region with consistent spontaneous strain direction, can be switched by applying external stress, endowing ferroelastic materials with promising potentials in mechanical switch, shape memory, signal processing, and superelasticity [4–7]. Since the first complete elastic hysteresis loop of Pb3PO4 was observed in 1976 [8,9], many pure inorganic ferroelastics (such as BiVO4 [10], Gd2(MoO4)3 [11] and PbZr1−xTixO3 [12]) have be studied on their application potentials [13,14]. Nonetheless, most of them face the obstacles in the further applications because of some drawbacks such as containing harmful metals, high energy consumption, or difficulty in regulation [15–17]. Emerging organic-inorganic hybrid crystals are regarded as an effective alternative to conventional inorganic ferroelastics owing to their simple processing, better mechanical flexibility, and environmental friendliness [18–21]. However, to date, the research on organic-inorganic hybrid ferroelastics is still immature, and the design and synthesis of hybrid ferroelastics with high spontaneous strain is a new challenge for scientists [22–25].
Exhilaratingly, the A2BX4-type halide hybrid perovskites (A = organic ammonium, B = metal ion, and X = halogen) featuring two-dimensional (2D) inorganic layers separated by organic cations have attracted increasing attention due to its potential as ferroic and optoelectronic materials [26–30]. When such kind of crystals undergo structural phase transition, the deformation of inorganic layers and the order-disorder changes of organic cations are prone to spontaneous shear distortion of the crystal lattice, leading to a change of the crystallographic unit cell shape, which provides favorable conditions for designing ferroelastics [26,31,32]. For instance, we previously reported a layered hybrid perovskite halide, (C6H5(CH2)4NH3)2[CuCl4], which undergoes a ferroelastic phase transition with an Aizu’s notation of 4/mmmF2/m at 337 K [32]. Inoue et al. reported a halide hybrid perovskite, (C2H5NH3)2[FeCl4], exhibiting ferroelasticity at below 433 K and canted antiferromagnetism at below 98 K [31]. However, the hybrid ferroelastics based on layered perovskites are still rare, and many reported cases have not been further studied on their spontaneous strains.
Notably, substitution strategy has been proved to be an effective design strategy to tune the phase transitions in hybrid crystals [33–36]. For instance, replacing hydrogen atom in the organic cations by heavier halogens with greater electronegativity usually increases the potential energy barrier for swing or rotating the cations, hence increases the phase-transition temperatures [37–40]. Moreover, substitution strategy generally could reduce the structural symmetry for lower-temperature phase and then to bring about a ferroelastic phase transition [41,42]. We previously found that the F-substitution in a layered perovskite halide, (4-fluorophenethylaminium)2[CuCl4], reduces the structural symmetry of room-temperature phase from orthorhombic to monoclinic, hence brings about a mmmF2/m ferroelastic transition with a spontaneous strain of 0.011 [26]. Herein, as an extension, to enrich the layered perovskite ferroelastic crystals and to increase their spontaneous strains, we chose a parent compound, (phenethylaminium)2[CdCl4], which crystallizes in the C2cb space group at room temperature [43,44], and applied halogen substitution on the meta-position of its phenethylaminium cation to further reduce molecular symmetry. Our efforts have yielded three new layered organic-inorganic hybrid perovskites, (3-XC6H5CH2CH2NH3)2[CdCl4] (X = F (1), Cl (2) and Br (3)). 1–3 have isomorphic high-temperature phases (HTPs) belonging to space group I4/mmm but different room-temperature phases (RTPs) belonging to space groups P21/a, P1 and P21/a, respectively, all of which are symmetrically reduced when compared with the parent one (C2cb). The thermal stabilities, structural details, and the underlying ferroelastic transition mechanisms were comprehensively studied by means of thermogravimetric analyses (TGA), differential scanning calorimetry (DSC), dielectric measurement, variable-temperature single-crystal X-ray structural analyses, polarization microscopy, and Hirshfeld surface analyses.
Thermal analyses showed that 1–3 are stable up to approximately 470, 483 and 486 K under N2 atmosphere, respectively (Fig. S2 in Supporting information). As shown in the DSC curves in a temperature range of 330–450 K (Fig. 1), a pairs of sharp thermal anomalies indicating reversible one-step phase transitions were observed at 376/371 K (T1), 436/430 K (T2), and 421/411 K (T3) in a heating/cooling cycle for 1–3, respectively. The total entropy changes (∆S) during the phase transitions in the heating process were 45.6, 43.7 and 33.8 J mol−1 K−1 for 1–3, respectively. For convenience, the phases of 1–3 at above phase-transition temperatures are labeled as 1_HTP, 2_HTP and 3_HTP, while those at below phase-transition temperature are labeled as 1_RTP, 2_RTP and 3_RTP, respectively.
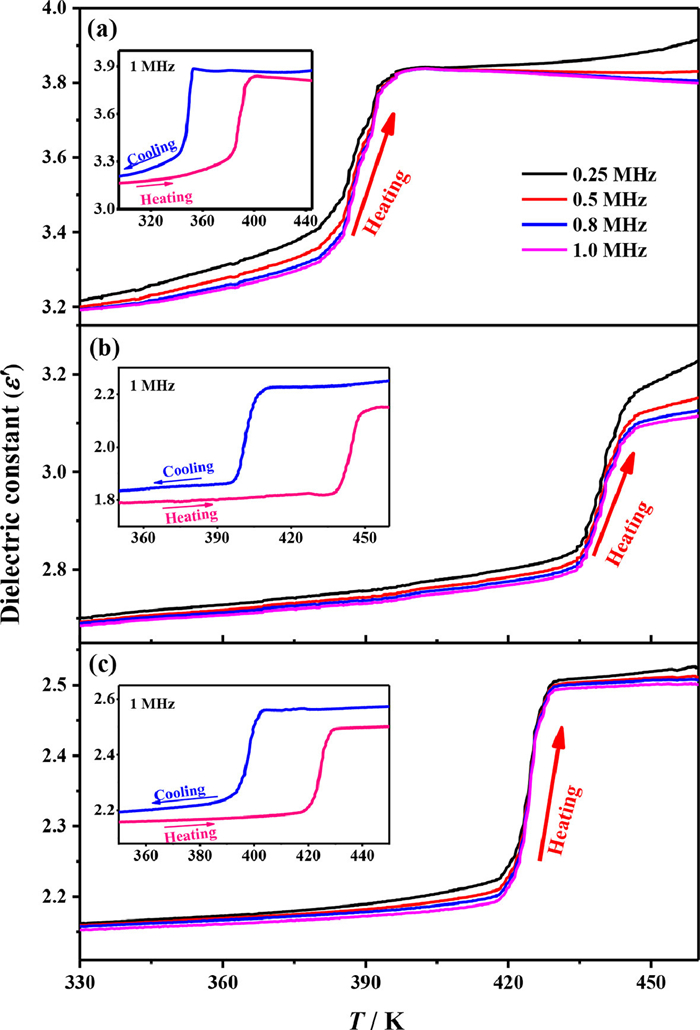
The temperature-dependent permittivities (ε = ε′ + iε′′) were measured on powder-pressed samples in the temperature range of 330–460 K at a rate of 4 K/min for 1–3. As shown in Fig. 2, all of them show step-like dielectric anomalies at around their phase-transition temperatures. In detail, the ε′ value (f = 1 MHz) of 1 is 3.2 at 330 K, then increases to 3.8 at 400 K with increasing temperature, the ε′ value (f = 1 MHz) of 2 is 1.8 at 350 K then increases to 2.2 at 460 K upon heating, and the ε′ value (f = 1 MHz) of 3 is 2.2 at 350 K then increases to 2.5 at 450 K upon heating. The reverse dielectric drops were observed in the following cooling runs (Fig. 2, inset). Such reversible step-like dielectric anomalies respond to the changes in the dynamic motions of the dipolar components, suggesting an order-disorder variation of the guest cations accompanying with phase transitions in 1–3.
To investigate the microscopic mechanism of the reversible phase transitions, the in-situ variable-temperature powder X-ray diffractions (Fig. S3 in Supporting information) and variable-temperature single-crystal X-ray diffractions were performed at 298 K, 386 K for 1, 293 K, 450 K for 2, and 298 K, 435 K for 3, respectively. The results revealed that all phases of 1–3 can be described as A2BX4-type layered perovskite structures. Each Cd2+ cation is coordinated with six Cl− ions to form a compressed octahedral coordination geometry with the equatorial and bridging Cd–Cleq bonds longer than the axial Cd–Clax bonds. Each CdCl6 octahedron links to four adjacent octahedra by sharing vertexes to form [CdCl4]∞ layers, which are separated by double layers of organic cations 3-XC6H5CH2CH2NH3+ (X = F, Cl and Br) with terminal –NH3 head orientating to the intra-layer cavities. Such a layered perovskite structure is stabilized by coulomb forces, N–H···Cl hydrogen bonds, and van der Waals forces between the organic cations and inorganic layers.
The structural analyses indicated that 1–3 have isomorphous HTPs but different RTPs. As shown in Fig. 3, the HTPs of 1–3 possess the same tetragonal space group I4/mmm (Z = 2) with similar cell parameters, and each of their asymmetric units include one organic cation and half [CdCl4]2− moieties. All organic cations are 8-fold crystallographically disordered about the 4-fold axes parallel to the c axis and the mirror perpendicular to the c axis. The Cd2+ ions locate at the original and body-centered positions in a regular compressed CdCl6 octahedral coordination.

For the RTPs of 1–3, their c-axis lengths are halved when compared with those in HTPs owing to the displacements of adjacent inorganic layers and the deviations of Cd2+ ions from the body-centered position (Fig. S4 in Supporting informaiton). When compared with the orthorhombic RTP (C2cb) in the parent (phenethylaminium)2[CdCl4], the structural symmetries of RTPs in 1–3 are reduced (Fig. S5 in Supporting informaiton). In detail, 1_RTP belongs to the monoclinic space group P21/a (non-standard setting of P21/c for convenience of structural comparison) with Z = 2, which is similar to the RTP of (C6H5(CH2)4NH3)2[CuCl4] [32]. In 1_RTP, each 3-FC6H5CH2CH2NH3+ cation freezes into 2-fold disordered about general position with occupancy of 0.55:0.45. For 2_RTP, it adopts lower-symmetry triclinic P1 space group with Z = 4, in which four crystallographically independent 3-ClC6H5CH2CH2NH3+ cations are frozen into an ordered state with slightly different orientations/conformations (Fig. S6 in Supporting informaiton). Accordingly, a greater distortion in the inorganic framework was observed in 2_RTP (Fig. S7 in Supporting informaiton). For 3_RTP, it also crystallizes in the monoclinic space group P21/a but with an increased Z value of 4, as the a-axis [10.9801(4) Å] and b-axis lengths [10.6320(5) Å] are 
As indicated by the above structural analyses, the transitions from the tetragonal HTPs to the symmetrically reduced RTPs in 1–3 belongs to 94 ferroelastic transitions deduced by Aizu [45]. In detail, for both 1 and 3, the HTP → RTP transitions belong to ferroelastic transitions with an Aizu notation of 4/mmmF2/m, as the point group transforms from D4h to C2h accompanied by a holistic decrease of symmetric elements from 16 (E, 2C4, C2, 2C’2, 2C"2, i, 2S4, σh, 2σv, 2σd) to 4 (E, C2, i, σh). For 2 with a triclinic RTP, the variation in symmetry of 2_HTP → 2_RTP belongs to a ferroelastic transition with an Aizu notation of 4/mmmF1 and a much more decrease of symmetry elements from 16 to 2 (E, i).
To visualize the ferroelastic transitions for 1–3, the variable-temperature polarization microscopy was performed on single crystals perpendicular to their (001) planes to inspect possible ferroelastic transition (Fig. S8 in Supporting informaiton). As illustrated in Fig. 4, the single crystals for 1–3 reveal a monodomain state at 298 K, as no external strain was applied to the single crystals, and become multi-domain ferroelastic states in the vicinity of the phase-transition temperatures, then change to monodomain paraelastic state upon further heating to above phase-transition temperatures, and the multi-domain ferroelastic states could reappeared when the temperature was cooled down to below the phase-transition temperatures. Taking 1 as an example, when the temperature rose to the vicinity of 371 K, two types of distinguishable striped domains appeared and gradually grew to cover the whole ab plane, illustrating the domain walls inside which the spontaneous strain orderly aligns. In this process, some crystal crackles appeared due to large spontaneous strains during phase transitions (vide infra). With further heating to 381 K, the striped domains faded away and the monodomain morphology appeared. When the temperature was decreased to 375 K, numerous slender striated domains reappeared. Upon cooling to 364 K (1_RTP, ferroelastic), the multi-domains grew and remained. For 3, it revealed similar domain revolutions with 1 as both of them belong to 4/mmmF2/m ferroelastic species, but the multi-domain stripes in 3_RTP looks more complicate than that in 1_RTP because of the lower structural symmetry of 3_RTP. For 2 with a much lower symmetric triclinic ferroelastic phase, when the crystal was cooled from 438 K (paraelastic phase) to 432 K (ferroelastic phase), plentiful complex and dense domain walls appeared, and the multi-domain state was fully developed to haze the crystal. In short, these repeatable domain revolutions in the heating-cooling cycles well confirmed the reversible ferroelastic transitions for 1–3.

To study the spontaneous deformation of the unit cell and further to evaluated the ferroelastic spontaneous strains, the temperature-dependent lattice parameters were obtained by carrying out Pawley refinements on the in-situ variable-temperature PXRD patterns (Tables S4 and S5 in Supporting information) in temperature range of 323–403 K for 1, 409–454 K for 2, and 393–440 K for 3, respectively. The thermal expansion coefficients were accordingly calculated by utilizing PASCal program [46]. As listed in Table S6 (Supporting information), the volumes of 1–3 show different degrees of positive volume thermal expansion in RTPs and HTPs, i.e., +214 × 10−6 K−1 and +201 × 10−6 K−1 for 1, +404 × 10−6 K−1 and +284 × 10−6 K−1 for 2, +105 × 10−6 K−1 and +1112 × 10−6 K−1 for 3, respectively, reflecting the enhanced dynamic motions of organic cations upon heating in these phases. It is worth to note that the significantly larger positive thermal expansion in 3_HTP should be mainly attributed by its unusual large intralayer thermal expansion when compared with the cases in 1 and 2, arising from its highly disordered organic cation with much larger bromine atom. In Figs. S8–S10 (Supporting information), all the lattice parameters show a certain degree of jumps at the phase-transition temperatures. From RTPs to HTPs, the cell volumes are increased by 5.1%, 8.0% and 5.4% in 1–3, respectively. Such relatively large volume changes during the ferroelastic transitions are mainly contributed by the increases of their c-axes lengths by 6.3%, 8.4% and 8.6% in 1–3, respectively. In contrast, the changes in the a-axis and b-axis lengths are not more than 1%, 2% and 3% in 1–3, respectively. As shown in Fig. 3, such dramatic changes on the c axes and cell volumes are highly associated with the displacement of the inorganic layers caused by the changes on molecular dynamics of the 3-XC6H5CH2CH2NH3+ (X = F, Cl, Br) cations from the ordered and tilting states in RTPs to the highly disordered and upright states in HTPs. In addition, these dramatic changes are responsible for the aforementioned crystal cracks during the ferroelastic transitions.
Based on the changes of cell parameters during phase transitions, the ferroelastic spontaneous strains were calculated for 1–3. The cell parameters for paraelastic and ferroelastic phases at the phase-transition temperatures were deduced by extrapolating the fitting lines of their HTPs and RTPs, respectively (Table S5). The necessary matrices were applied to convert the cells of 1_HTP, 2_HTP and 3_HTP to match those of 1_RTP, 2_RTP and 3_RTP, respectively (for details, see Supporting information). The calculation yielded the total spontaneous strains (Ԑss) of 0.16, 0.13 and 0.12 for 1–3, respectively. These values are smaller than those observed in a few layered perovskite ferroelastics with smaller organic cations such as (C3H5NH3)2[CdCl4] (0.239) [47] and (C4H9N)2[PbBr4] (0.191) [48], but larger than those in most hybrid crystals with discrete and one-dimensional coordination units such as (Me3NCH2Br)2[CoBr4] (0.068) [49] and (Me4P)[Cd(SCN)3] (0.062) (Table S7 in Supporting information) [26]. Such relatively large spontaneous strains in 1–3 should be highly associated with the order-disorder dynamic transitions of the flexible organic cation and the displacement of adjacent inorganic layers.
Considering that 1–3 have almost same inorganic layers and isostructural HTPs, their distinct RTPs should be ascribed to the slight changes on the halogen-involving intermolecular interactions. The Hirshfeld surface analyses [50] were performed on the 3-XC6H5CH2CH2NH3+ (X = F, Cl and Br) cations in 1_RTP, 2_RTP and 3_RTP, respectively, to comprehensively investigate their different halogen-involving intermolecular interactions. In detail, the Hirshfeld surfaces are displayed in a red-white-blue scheme, indicating the intermolecular contacts shorter (red), around (white), and longer (blue) than van der Waals separation. In Fig. 5, the H···Cl contacts associating with 21.1%, 30.6%−30.9%, and 20.7%−22.8% of the surface areas for 1–3, respectively, together with the coulomb interactions, make a major contribution to attractive host-guest molecular interactions between inorganic layers and organic cations. Conversely, the H···H contacts associating with 47.4%, 35.2%−37.4%, and 35.8%−37.3% of the surface areas for 1–3, respectively, together with the repulsive coulomb interactions, make the main contribution guest-guest repulsive interactions for adjacent cations. Notably, for the adjacent cations, the X···H (X = F, Cl and Br) contacts associating with 20.9%, 12.0%−12.5%, and 20.2%−24.2% of the surface area contribute important attractive interactions for adjacent cations in 1–3, respectively. The minimum (di, de) pairs of intermolecular short contact indicate that the strongest contacts are (0.88 Å, 1.5 Å) in 1_RTP, (0.80 Å, 1.40 Å) in 2_RTP, and (0.82 Å, 1.42 Å) in 3_RTP, respectively, being associated with N−H···Cl weak hydrogen bonds.
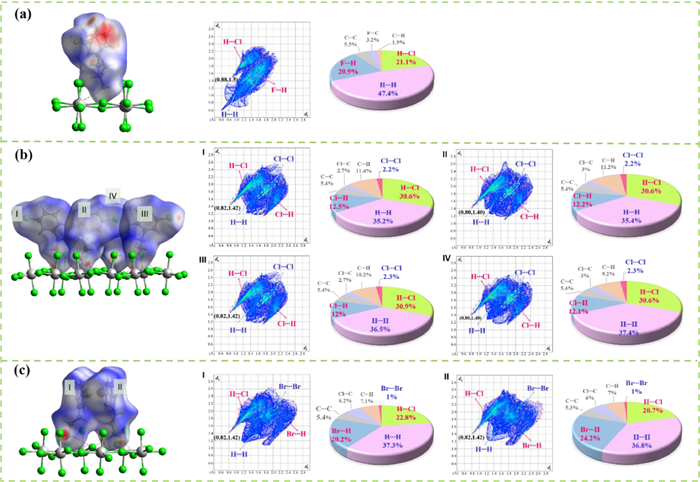
According to the increased atom weights from F to Cl and then Br, one could expect that the order of phase-transition temperatures is T1 < T2 < T3, if taking account the dispersion forces as the main intermolecular interactions. However, the experimentally observed T2 is higher than T3. Such an exceptional case should be ascribed to the much stronger host-guest attractions in 2, as indicated by its higher proportion of H···Cl (30.6%–30.9%) contacts than those in 1 (21.1%) and 3 (20.7%–28.8%). Namely, with the relatively stronger host-guest hydrogen bonds (Table S3 in Supporting information), the organic cation 3-ClC6H5CH2CH2NH3+ in 2 needs to overcome a higher energy barrier to undergo order-disorder transition, hence gives a higher phase-transition temperature. Besides that, the weak halogen···halogen interactions, i.e., Cl···Cl (2.2%–2.3%) in 2 and Br···Br (1.0%) in 3, also make additional contributions to establish the higher energy barriers for rotating the organic cations and to give higher phase-transition temperatures for 2 and 3.
Compared with the orthorhombic RTP (C2cb) in parent (C6H5CH2CH2NH3)2[CdCl4], the RTPs in 1–3 have reduced crystallographic symmetries, as the 3-XC6H5CH2CH2NH3+ (X = F, Cl, Br) cations with reduced molecular symmetry freeze into an ordered state and being tilting relative to the inorganic layers at room temperature, giving monoclinic or triclinic RTPs for 1–3. In particular, with the stronger host-guest interactions, the 3-ClC6H5CH2CH2NH3+ cations in 2_RTP seem not easy to change their conformations hence are frozen into similar but nonidentical conformations for balancing abundant weak interactions, and eventually give the triclinic space group with Z = 4 for 2_RTP. By contrast, the smaller 3-FC6H5CH2CH2NH3+ cations in 1 can adjust the conformations relatively easily, thus can not be completely frozen at room temperature, hence give a relatively simple monoclinic structure (P21/a) with Z = 2. While the 3-BrC6H5CH2CH2NH3+ cations in 3 are completely frozen into an ordered state and shows two different conformations in the asymmetric unit (P21/a, Z = 4), i.e., giving a decreased crystallographic symmetry than 1 (P21/a, Z = 2). In short, the delicate changes caused by the different halogen-involving intermolecular interactions play important roles on tuning the phase-transition temperatures and yielding the distinct crystal symmetry of ferroelastic phases for 1–3. Such influences on phase transitions well reflect the delicate and complex balances established by abundant weak intermolecular interactions in layered perovskites.
In summary, by employing halogen-substituted 3-XC6H5CH2CH2NH3+ (X = F, Cl and Br) organic cation, three new A2BX4-type layered hybrid perovskites, i.e., 1–3, were synthesized. They undergo ferroelastic phase transitions at 376/371 K, 436/430 K, and 421/411 K, respectively, between the same tetragonal HTPs (I4/mmm, Z = 2) and different RTPs with the space groups of P21/a (Z = 2), P1 (Z = 4), and P21/a (Z = 4), respectively, accompanying with spontaneous strains of 0.16, 0.13 and 0.12, respectively. Based on the single-crystal structures, the Hirshfeld surface analyses disclosed that the different halogen-involving intermolecular interactions play crucial roles on tuning the phase-transition temperatures, and more importantly, on controlling the conformations of frozen organic cations to establish their distinct RTPs. This work demonstrates a promising strategy to obtain new reversible ferroelasticity taking advantage of halogen regulation, and provides important clues to enrich the layered perovskite family for exploring new advanced functional materials based on hybrid crystals.
During our submission, the ferroelastic phase transition of 1 was briefly reported by Yue et al. [51]. We note that we have comprehensively investigated the spontaneous strains during the ferroelastic phase transition for 1–3, and more importantly, discussed the effect of different halogen substitutions on their phase transitions.
The authors declare that they have no conflict of interest.
This work was supported by National Natural Science Foundation of China (NSFC, Nos. 22071273 and 21821003), and Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (No. 2017BT01C161).
Supplementary material associated with this article can be found, in the online version, at doi:
U. Bismayer, E. Salje, A.M. Glazer, et al., Phase Transit. 6 (1986) 129–151. doi: 10.1080/01411598608221634
S.A.T. Salje, E. Salje, J. Phys. C Solid State Phys. 21 (1988) 277–285. doi: 10.1088/0022-3719/21/2/011
J. Sapriel, Phys. Rev. B 12 (1975) 5128–5140. doi: 10.1103/PhysRevB.12.5128
E.G. Fesenko, V.G. Gavrilyatchenko, A.F. Semenchev, Ferroelectrics 100 (1989) 195–207. doi: 10.1080/00150198908007915
J. Li, Y. Zhu, P.Z. Huang, et al., Chem. Eur. J. (2022) e202201005.
D.X. Liu, Z.H. Yu, X.X. Chen, et al., Chin. Chem. Lett. 34 (2023) 107310. doi: 10.1016/j.cclet.2022.03.033
H.Y. Zhang, C.L. Hu, Z.B. Hu, et al., J. Am. Chem. Soc. 142 (2020) 3240–3245. doi: 10.1021/jacs.9b13446
E.K.H. Salje, G. Hoppmann, Mat. Res. Bull. 11 (1976) 1545–1550. doi: 10.1016/0025-5408(76)90107-0
E.K.H. Salje, Ferroelectrics 104 (1990) 111–120. doi: 10.1080/00150199008223816
P.J. Fillingham, J. Appl. Phys. 38 (1967) 4823–4829. doi: 10.1063/1.1709228
K. Aizu, A. Kumada, H. Yumoto, et al., J. Phys. Soc. Jpn. 27 (1969) 511. doi: 10.1143/JPSJ.27.511
Z. An, S. Xie, N. Zhang, et al., APL Mater. 9 (2021) 030702. doi: 10.1063/5.0038996
V. Nagarajan, A. Roytburd, A. Stanishevsky, et al., Nat. Mater. 2 (2003) 43–47. doi: 10.1038/nmat800
K. Dorr, Nat. Mater. 15 (2016) 497–498. doi: 10.1038/nmat4596
T. Delgado, A. Tissot, L. Guenee, et al., J. Am. Chem. Soc. 140 (2018) 12870–12876. doi: 10.1021/jacs.8b06042
C. Su, M. Lun, Y. Chen, et al., CCS Chem. 4 (2022) 2009–2019. doi: 10.31635/ccschem.021.202101073
H.Y. Zhang, Y.Y. Tang, P.P. Shi, et al., Acc. Chem. Res. 52 (2019) 1928–1938. doi: 10.1021/acs.accounts.8b00677
X.X. Chen, X.Y. Zhang, D.X. Liu, et al., Chem. Sci. 12 (2021) 8713–8721. doi: 10.1039/D1SC01345A
D.W. Fu, H.L. Cai, Y. Liu, et al., Science 339 (2013) 425–428. doi: 10.1126/science.1229675
J.X. Gao, X.N. Hua, P.F. Li, et al., J. Phys. Chem. C 122 (2018) 23111–23116. doi: 10.1021/acs.jpcc.8b07853
X. Meng, Z.B. Liu, K. Xu, et al., Inorg. Chem. Front. 9 (2022) 1603–1608. doi: 10.1039/D1QI01533H
X. Xiao, J. Zhou, K. Song, et al., Nat. Commun. 12 (2021) 1332. doi: 10.1038/s41467-021-21493-w
Z.B. Liu, L. He, P.P. Shi, et al., J. Phys. Chem. Lett. 11 (2020) 7960–7965. doi: 10.1021/acs.jpclett.0c02235
M. Rok, B. Zarychta, R. Janicki, et al., Inorg. Chem. 61 (2022) 5626–5636. doi: 10.1021/acs.inorgchem.2c00363
X.Q. Xu, H. Zhang, X.Q. Huang, et al., Inorg. Chem. Front. 8 (2021) 1197–1204.
Y.P. Gong, X.X. Chen, G.Z. Huang, et al., J. Mater. Chem. C 10 (2022) 5482–5488. doi: 10.1039/D2TC00335J
B. Huang, L.Y. Sun, S.S. Wang, et al., Chem. Commun. 53 (2017) 5764–5766. doi: 10.1039/C7CC02408H
B. Huang, J.Y. Zhang, R.K. Huang, et al., Chem. Sci. 9 (2018) 7413–7418. doi: 10.1039/C8SC02917B
P.P. Shi, S.Q. Lu, X.J. Song, et al., J. Am. Chem. Soc. 141 (2019) 18334–18340. doi: 10.1021/jacs.9b10048
J. Li, X. Liu, P. Cui, et al., Sci. China Chem. 62 (2019) 1257–1262.
Y. Nakayama, S. Nishihara, K. Inoue, et al., Angew. Chem. Int. Ed. 56 (2017) 9367–9370. doi: 10.1002/anie.201703898
B. Huang, B.Y. Wang, Z.Y. Du, et al., J. Mater. Chem. C 4 (2016) 8704–8710. doi: 10.1039/C6TC02715F
Y.Y. Yu, P.Z. Huang, Y.Z. Wang, et al., Chin. Chem. Lett. 32 (2021) 3558–3561. doi: 10.1016/j.cclet.2021.02.040
H. Ye, W.H. Hu, W.J. Xu, et al., APL Mater. 9 (2021) 031102. doi: 10.1063/5.0035793
Y.J. Cao, L. Zhou, L. He, et al., Chem. Eur. J. 26 (2020) 14124–14129. doi: 10.1002/chem.202001266
Y. Yu, P. Huang, Y. Wang, et al., Chin. Chem. Lett. 32 (2021) 3558–3561. doi: 10.1016/j.cclet.2021.02.040
S. Liu, L. He, Y. Wang, et al., Chin. Chem. Lett. 33 (2022) 1032–1036. doi: 10.1016/j.cclet.2021.07.039
X.K. Liu, F. Gao, J. Phys. Chem. Lett. 9 (2018) 2251–2258. doi: 10.1021/acs.jpclett.8b00755
T. Schmitt, S. Bourelle, N. Tye, et al., J. Am. Chem. Soc. 142 (2020) 5060–5067. doi: 10.1021/jacs.9b11809
M. Li, M.S. Molokeev, J. Zhao, et al., Adv. Opt. Mater. 8 (2020) 2000418. doi: 10.1002/adom.202000418
H.Y. Liu, H.Y. Zhang, X.G. Chen, et al., J. Am. Chem. Soc. 142 (2020) 15205–15218. doi: 10.1021/jacs.0c07055
W. Ning, F. Gao, Adv. Mater. 31 (2019) e1900326. doi: 10.1002/adma.201900326
M. Groh, R. Spengler, H. Burzlaff, et al., Acta Crystallogr. Sect. C: Cryst. Struct. Commun. 53 (1997) 1199–1201. doi: 10.1107/S0108270197005726
S. Kassou, A. Kaiba, P. Guionneau, et al., J. Struct. Chem. 57 (2016) 737–743. doi: 10.1134/S0022476616040168
K. Aizu, J. Phys. Soc. Jpn. 27 (1969) 387–396. doi: 10.1143/JPSJ.27.387
M.J. Cliffe, A.L. Goodwin, J. Appl. Crystallogr. 45 (2012) 1321–1329. doi: 10.1107/S0021889812043026
S. Han, X. Liu, J. Zhang, et al., J. Mater. Chem. C 6 (2018) 10327–10331. doi: 10.1039/C8TC03517B
Z.X. Wang, W.Q. Liao, H.Y. Ye, et al., Dalton Trans. 44 (2015) 20406–20412. doi: 10.1039/C5DT03277F
X.N. Hua, C.R. Huang, J.X. Gao, et al., Dalton Trans. 47 (2018) 6218–6224. doi: 10.1039/C8DT00786A
M.A. Spackman, D. Jayatilaka, CrystEngComm 11 (2009) 19–32. doi: 10.1039/B818330A
Z.Y. Yue, W. Luo, N. Wang, et al., CrystEngComm 25 (2023) 1270–1275. doi: 10.1039/D2CE01390H

Figure 2 Temperature-dependent dielectric constants (ε′) measured at 0.25-1.0 MHz upon heating for (a) 1, (b) 2 and (c) 3. Inset: temperature-dependent ε′ at 1 MHz during a heating-cooling cycle.

Figure 3 The crystal packing structures of (a) 1, (b) 2 and (c) 3 at RTP and HTP, respectively.

Figure 4 Evolution of the domain structure of (a) 1, (b) 2 and (c) 3 in the heating-cooling cycle.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
