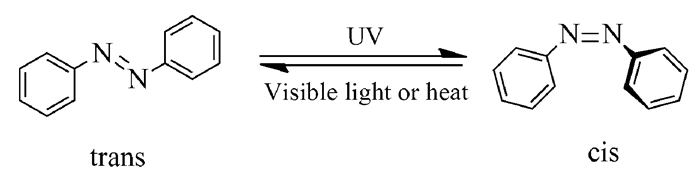
图式 1.
偶氮苯的光致异构化
Scheme 1.
Photoisomerization of azobenzene

早在1937年,英国化学家Hartley就发现偶氮苯的饱和溶液暴露在光照下时,其可见光吸收发生了变化[1],进而发现偶氮苯分子具有两种几何构型。随着偶氮苯分子研究的进一步展开,人们发现偶氮苯分子在紫外光照射下会发生反式到顺式的构型转变,而在可见光照射或者加热条件下则重新从顺式变为反式构型(图式 1)。
反式构型偶氮苯的基态能级要比顺式构型偶氮苯的基态能级低50kJ/mol,所以反式构型的偶氮苯稳定性要优于顺式构型的偶氮苯,这也使得顺式构型的偶氮苯既可以在低能量的可见光照(λ~450nm)条件下发生光异构化,也可以在避光条件下经过热作用自发回到反式构型,而反式构型的偶氮苯则需要在高能量的紫外光照(λ~360nm)条件下才会转变为顺式构型。偶氮苯分子可以在顺式和反式两个异构体间进行可逆的转换。
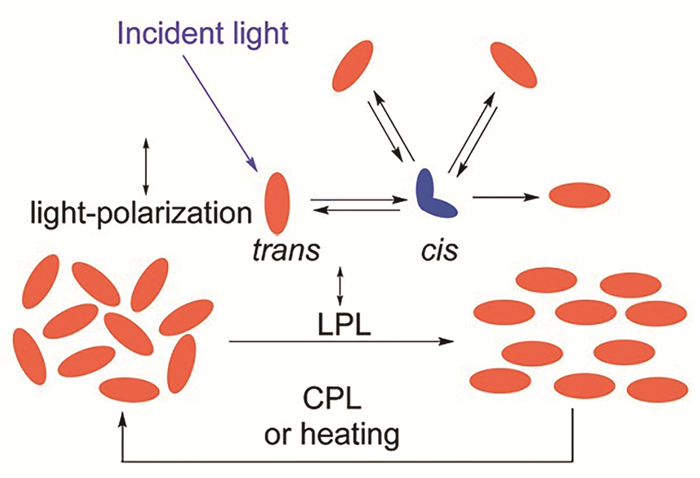
偶氮苯分子具有棒状结构,并且具有一定的刚性,可以作为液晶基元。偶氮苯基元在液晶表面排列时,如果受到偏振光的照射,由于偶氮苯分子的光响应异构化,会导致液晶在整体上发生光致取向。这是由于偶氮苯分子更倾向于吸收偏振方向与其长轴方向一致的光,而不会吸收偏振方向与分子长轴方向垂直的光。偏振光照射条件下,当偶氮苯的反式构型异构化为顺式构型时,由于偶氮苯顺式构型比反式构型的稳定性差,光照下顺式构型的偶氮苯分子会自发地返回反式构型,所以偶氮苯分子在光照条件下顺式-反式构型不断的异构。当偶氮苯分子从顺式构型异构回到反式构型时,分子长轴方向是随机的,只有当分子长轴与偏振光的偏振方向相互垂直,偶氮苯分子才不会再发生异构。所以使用线性偏振光(Linearly polarized light)照射时,偶氮苯分子倾向于垂直光偏振方向。而圆偏振光(Circularly polarized light)照射或加热则会无规取向(图 1)[2]。
将偶氮苯与高分子相结合的方法主要有三种[2],第一种方法是直接将小分子偶氮苯与高分子基质进行掺杂,这种方法虽然简单,但是偶氮苯分子很容易发生自聚集而导致相分离,并且这种方式得到的光响应体系均匀性和稳定性也较差[3];第二种方法是通过非共价作用,将偶氮苯与其他分子组装成偶氮苯超分子体系,这种方法避免了偶氮苯的自聚集,而且稳定性有一定的提高;第三种方法是通过化学反应,将偶氮苯连接到高分子的主链或者侧链上,该方法往往需要复杂的合成过程,但是体系稳定性较好,因此应用较为广泛。
偶氮苯分子的光致异构会引起材料的化学结构、空间分布及聚集状态的变化,因此偶氮苯的光致异构化转变会引起高分子结构或者构象的转变[4],从而使得高分子对光照具有一定的响应性能。在液晶偶氮苯高分子中,由于偶氮苯的光致取向特性,导致液晶分子的定向排列发生改变,从而引起液晶的相转变。1995年,Ikeda等[5]报道了偶氮苯液晶高分子在光照条件下发生向列相液晶到各向同性聚合物转变的现象。除了液晶高分子的相变,2012年,Akiyama等[6]将偶氮苯基团通过共价作用连接到小分子糖醇上实现了小分子偶氮苯的光致固液转变。由于偶氮苯高分子链构象复杂,不易形成结晶以及空间位阻较大等原因,而难以表现出类似于小分子偶氮苯的光致固液转变特性。2017年,笔者课题组将偶氮苯键接到丙烯酸分子中,制备了丙烯酸类偶氮苯单体,并通过可控活性自由基聚合方法制备了具有光致可逆固液转变的偶氮苯高分子[7]。更重要的是我们证明了偶氮苯高分子材料的光致液化现象是由偶氮苯的光致异构化改变了高分子的玻璃化转变温度(Tg)引起的,并将该偶氮苯高分子成功的应用于可修复涂层、降低表面粗糙度和转印技术中。其后,根据偶氮苯高分子独特的光致固液转变性质,我们开展了一定的基础研究和实际应用[8~10]。
本文将主要介绍丙烯酸酯类偶氮苯高分子的合成,分析该类高分子的光致固液转变的原理,并介绍基于此机理开发的偶氮苯高分子光控胶粘剂、光致动器、光致热导开关器件以及非热纳米压印。
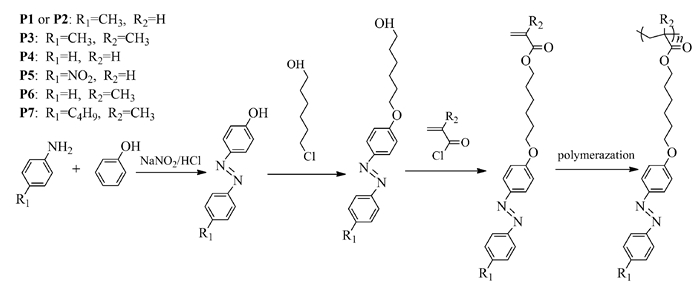
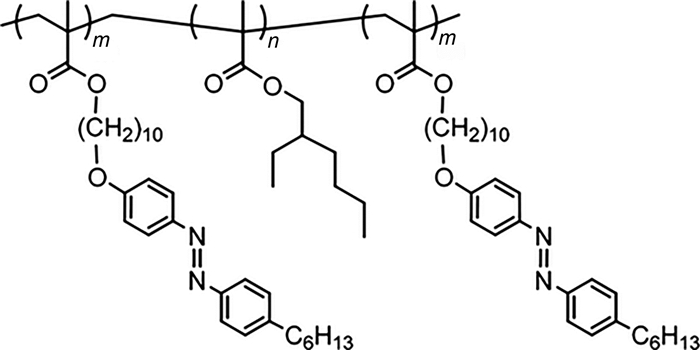
(甲基)丙烯酸酯类高分子作为重要的高分子种类,具有易于制备和改性的特点,所以通常通过(甲基)丙烯酸酯将偶氮苯分子结构引入到高分子材料中。因为传统的自由基聚合难以控制高分子的分子量以及分子量分布,所以有必要通过活性自由基聚合方法制备偶氮苯高分子。研究表明,甲基丙烯酸酯类偶氮苯高分子可以通过常用的原子转移自由基聚合(ATRP)以及可逆加成断裂链转移聚合(RAFT)方法制备[11, 12]。但是有报道认为,ATRP难以控制丙烯酸酯类偶氮苯单体的聚合[13]。对于这一问题,Ito等[14, 15]报道了使用ATRP方法制备丙烯酸酯类偶氮苯高分子均聚物和嵌段共聚物;笔者等[7, 8]先后成功使用RAFT和ATRP方法制备了丙烯酸酯类偶氮苯高分子。具体合成路线如图式 2所示。
该高分子的单体通过3步反应合成,高分子P1~P7可以通过RAFT方法制备,其中链转移剂为2-氰基-2丙基苯并二硫(CPDB),引发剂为偶氮二异丁腈(AIBN),溶剂为苯甲醚。高分子量的偶氮苯高分子P1还可以通过ATRP方法制备,使用五甲基二乙烯三胺(PMDETA)为配体,铜和溴化铜为催化剂,α-溴代苯乙酸甲酯(MBPA)为引发剂,溶剂为苯甲醚。
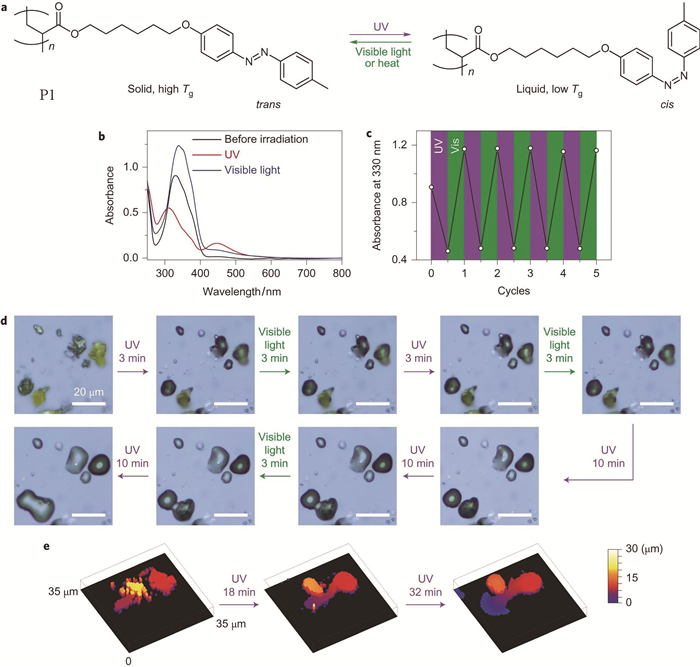
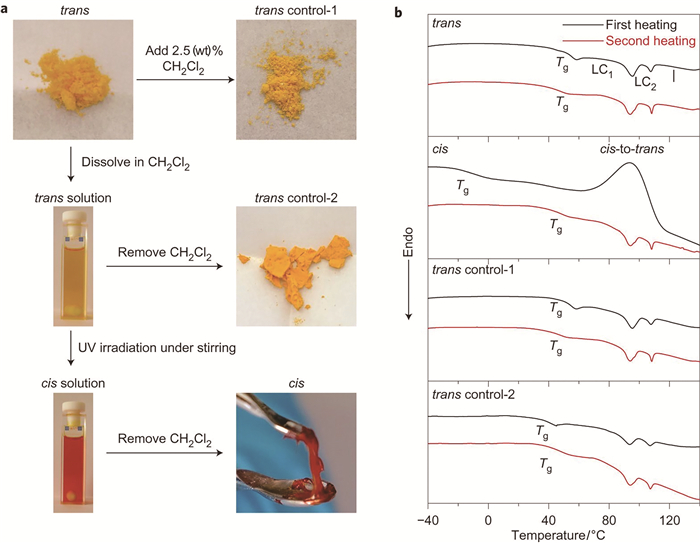
为了研究光诱导丙烯酸酯类偶氮苯高分子固液转变现象的机理,我们设计了代表性的丙烯酸酯类偶氮苯高分子P1[7]。首先,在宏观尺度上验证了P1分子的可逆固液转变现象,紫外光和可见光照射分别可以诱导P1偶氮苯高分子进行反式-顺式和顺式-反式的异构化转变。光照之前,图 2(a)的UV-Vis谱显示,该分子的反式构型在330nm处有强的π-π*跃迁吸收峰;紫外光照射后,该处跃迁吸收变弱,并且在450nm处的n-π*跃迁增强,表明偶氮苯基团进行了反式-顺式异构化转变。该光致异构化过程至少可重复5次(图 2(c))。与此同时,光学显微镜图片显示,紫外光(365nm,67mW·cm-2,1min)辐照可以使样品由粉末状态转变为液滴状态,而随后的可见光照射(530nm,0.2mW·cm-2,1min)则并不能改变液滴的形貌(图 2(d))。共聚焦显微镜图片显示,偶氮苯高分子样品粉末在紫外光(365nm,67mW·cm-2)照射后,高度变低,而且与表面的接触面积增大(图 2(e))。这些实验都证明P1分子在紫外光照射后可以由固体转变为液体。
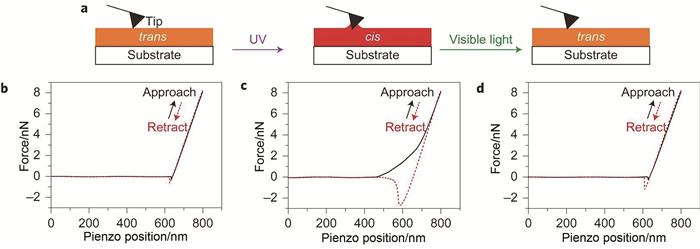
在微观尺度上,为了进一步证明该固液转变现象是可逆的,使用扫描力显微镜研究了偶氮苯高分子P1膜表面的力学性能(图 3(a))。当原子力针尖与反式构型偶氮苯高分子旋涂膜接触时,力随着位置线性增加,则表明膜为固体(图 3(b))。在紫外光照射后,偶氮苯高分子膜发生了反式构型向顺式构型的转变,粘附力显著增加到了2.7nN(图 3(c)),证实顺式构型比反式构型偶氮苯高分子的粘性更大。这是因为针尖接触顺式构型偶氮苯高分子的液体表面时,使得液面发生弯曲所产生的毛细作用力远大于反式偶氮苯高分子表面与针尖间的范德华力。在可见光照射后,力-位置曲线与紫外光照射前一致,说明顺式构型偶氮苯高分子液体转变为反式构型偶氮苯高分子固体,并且偶氮苯高分子P1的固液、液固转变是可逆的(图 3(d))。
使用差示扫描量热仪(DSC)分别测量了固液转变前后偶氮苯高分子的Tg。为了得到足量用于测试的顺式构型偶氮苯高分子样品,先将反式样品溶解于二氯甲烷中,然后在不断搅拌条件下,使用紫外光照射溶液,将新生成的顺式样品的溶液在常温条件下旋干,之后立即转移至DSC中进行测试。图 4(b)显示,顺式偶氮苯高分子的Tg(-10℃)明显低于反式偶氮苯高分子的Tg(48℃)。对比实验证明,在最大强度紫外光(68.8mW·cm-2)照射下,P1表面的温度不超过35℃,即上述固液转变过程并不是紫外光照射的光热效应所导致。流变测试表明,在可见光照射前,顺式构型偶氮苯高分子损耗角正切tanδ大于1,表现为液体性能;可见光照射后,反式构型的偶氮苯高分子tanδ小于0.15,具有固体性能。说明可见光照射的偶氮苯高分子发生了液固转变。经上述实验证明,反式-顺式异构化导致了偶氮苯高分子的Tg降低,从而实现了偶氮苯高分子的固液转变。
另外设计了丙烯酸酯类偶氮苯高分子P3~P5,实验结果表明,顺式P3、P4的Tg低于室温,紫外光照射条件下均发生了固液转变,说明该类偶氮苯高分子光致异构化是一种普适的现象。P5含有推-拉型偶氮苯[16],该类偶氮苯基团的偶极矩过大、半衰期短,所以该类偶氮苯分子不适于制备固液转变材料。上述基础研究证明,半衰期长且玻璃化转变温度低于室温的顺式结构偶氮苯高分子可以通过光致异构化调控Tg,并且在室温条件下实现可逆的光致固液转变。
Akiyama等[17]研究发现,具有短的烷基取代基以及短的亚甲基间隔基的偶氮高分子难以通过紫外光照液化。笔者等[18]进一步研究发现,含有短的(0个和2个)和长的(20个)亚甲基间隔基的丙烯酸酯类偶氮苯分子均难以实现固液转变。
因此,对于线性偶氮苯高分子,侧链为偶氮苯型(偶氮苯基团无强推拉电子取代基)的高分子,有利于光致固液转变。偶氮苯高分子具有合适的烷基取代基以及间隔基的长度,对实现室温下的光致固液转变具有重要的意义。
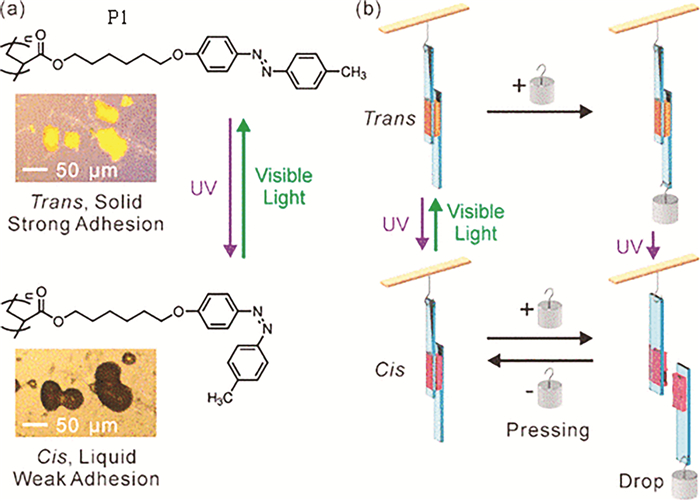
我们使用RAFT聚合方法制备了偶氮苯高分子P1,在室温条件下,反式构型偶氮苯高分子为固体状态,具有强的粘附力;紫外光照后,顺式构型的偶氮苯高分子为液体状态,具有弱的粘附力;可见光照射后偶氮苯高分子发生可逆的液固转变,因此P1可以用于光控粘合剂材料(图 5(a))。固态的P1没有足够的链迁移率,可以将两个基底牢固结合,当使用紫外光照射时,高分子P1变为液态后,促进了两个基底分离(示意图 5(b))[9]。
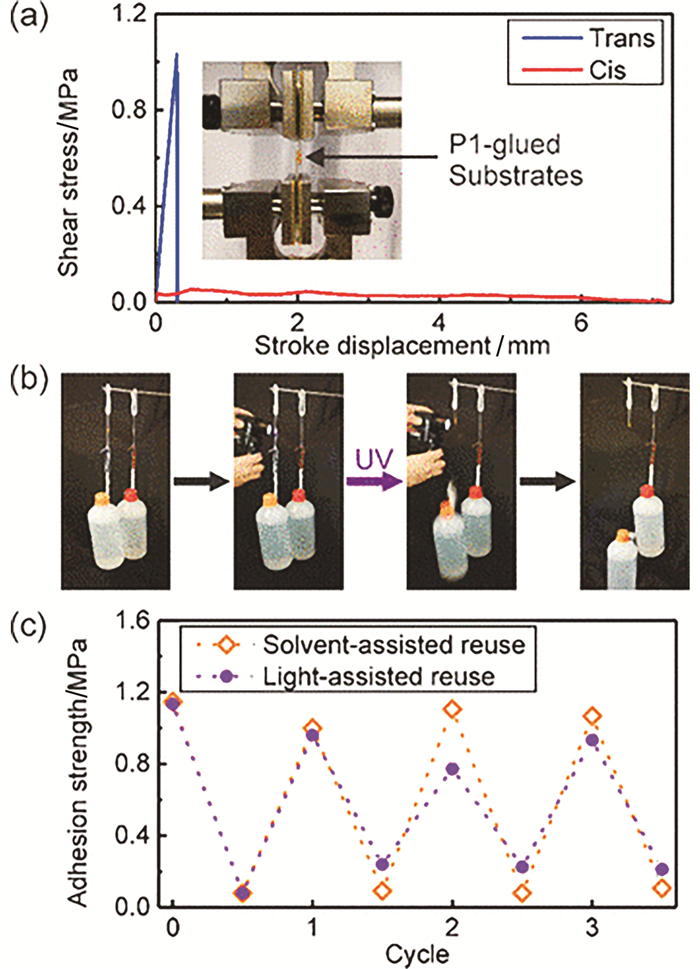
通过拉伸设备(图 6(a)中的插图)测试可知,反式和顺式构型偶氮苯高分子的粘结强度分别为1.02和0.08 MPa(图 6(a))。当两个石英基底的粘结面积为0.375cm-2时,足以吊起一瓶水的重量(图 6(b))。P1在溶剂(图 6(c),紫色曲线)和光(图 6(c),橙色曲线)辅助下的重复使用性能如图 6(c)所示,在分别进行的3个光异构化循环测试中的粘结强度为其初始状态的68%和87%。
Akiyama等[15, 19, 20]以ABA型三嵌段偶氮共聚物(图式 3)作为光控粘合剂,在对紫外线透明的丙烯酸酯基板上研究了光异构化过程,证明了紫外线照射会导致粘合强度降低,可见光照射会导致粘合强度增加。进一步研究表明,乙醇能够除去基材表面的粘合剂[15],并且约50%偶氮嵌段含量时可以使粘合效果以及清除效果达到最佳[19]。
对于光控偶氮苯粘合剂,其优点在于无污染、可重复利用,同时光具有高时空分辨率,可以精确地控制粘附与脱附。以光诱导的可逆的固液转变为设计胶粘剂的基础,将有效改善粘合剂的脱粘以及拓展粘合剂的应用范围。
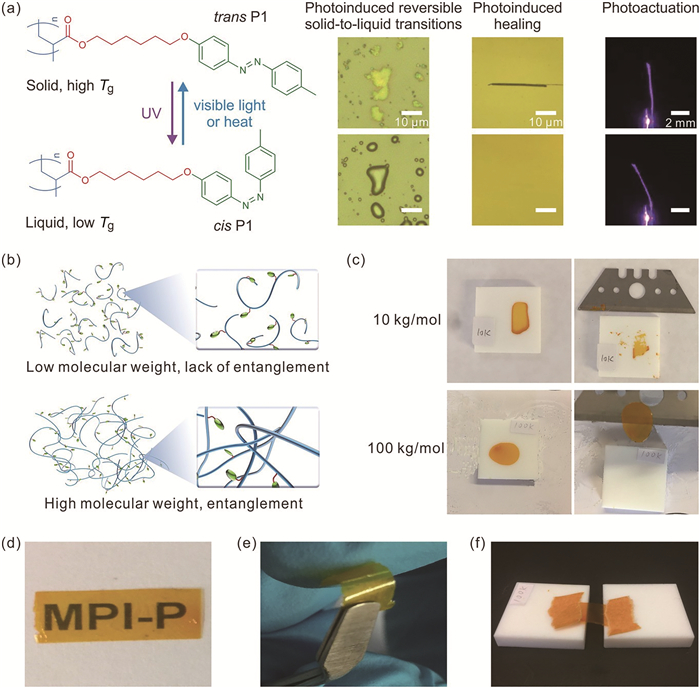
我们制备了高分子量的偶氮苯高分子P1,其能够可逆的发生固液转变以及自修复。通过滴膜的方法制备了可以弯曲的偶氮苯高分子非支撑膜(图 7(a))[8]。实验发现,分子量为10~53 kg/mol的高分子膜在剥离过程中发生了破裂(图 7(c));而高分子量(80与100 kg/mol)的膜则可以剥离。这是由于低分子量的P1缠结较少,高分子量的P1链缠结较多(图 7(b))。使用Wool高分子链缠结模型[21]通过理论计算证明,该高分子的临界缠结分子量约为68kg/mol。缺乏缠结的低分子量偶氮苯高分子坚硬易碎,而缠结的高分子量偶氮苯高分子P1-100k坚韧且具有良好的可加工性能(图 7(e~f))。
为了进一步研究P1-100k薄膜的机械响应性能,首先制备了拉伸偶氮苯高分子薄膜。薄膜拉伸后偶氮苯液晶基元沿着拉伸方向排列(图 8(a)),在紫外光光照10min后,薄膜靠近光源,弯曲角度为62°;使用蓝光照射后,薄膜远离光源弯曲,50s后恢复到初始的垂直位置。研究未拉伸的偶氮苯薄膜表明(图 8(b)),薄膜在紫外光照10min后,同样靠近光源,弯曲角度为26°,小于拉伸后薄膜的弯曲角度;蓝光照射10min后,则可以恢复到初始的垂直位置。不同于传统的交联型液晶弹性体,该偶氮苯高分子薄膜是通过高分子链缠结作为物理交联点而形成液晶弹性体,并不是通过化学交联,这种交联类型的特点是薄膜可以通过溶液重塑成型。此研究同时证明薄膜的弯曲并不需要预先进行液晶基元的取向。

研究证明,低分子量的P1(Mn=9.9kg·mol-1)和P2(Mn=27kg·mol-1)偶氮苯高分子具有光致可逆固液转变现象[9, 22]。反式构型P1-100k的Tg(≈80℃)高于室温,因此是固体;顺式构型P1-100k的Tg(≈7℃)低于室温,是液体,即高分子量偶氮苯高分子P1-100k也具有固液转变现象。另外,通过实验证明,在紫外光照射下,P1-100k的粉末从各向同性固体变为各向同性液体,光致固液转变过程不涉及液晶相到各向同性的转变。光诱导的从高Tg到低Tg的转变在P1-100k各向同性未拉伸膜中产生了约0.92N·m-1的表面应力变化,这可能会导致非支撑膜的光诱导弯曲。在紫外光照射(10min)过程中,由于高Tg向低Tg的过渡,膜表面发生液化而变得越来越柔软,从而更容易弯曲。
该研究还基于光致固液转变现象实现了致动器的修复(图 9(a))。P1-100k致动器的局部划痕经紫外光(365nm,51mW·cm-2,20min)照射,使偶氮苯高分子液化并发生流动,最后导致划痕消失(图 9(b));然后,经可见光(470nm,9mW·cm-2,20min)照射将愈合的P1-100k固化,完成修复过程。当新的划痕出现时,可以再次进行光修复。另外,将致动器切成两半(图 9(c))后,使用紫外光(20min,365nm,51mW·cm-2)照射,然后将经紫外光照射的侧面压在一起,并使用蓝光(20min,470nm,9mW·cm-2)固化后,薄膜仍然具有光致弯曲现象(图 9(d))。利用光致固液转变的机理,还可以对偶氮苯高分子光致动器进行微结构修饰。这种具有微结构的光致动器在运输设备、微致动器、光控摩擦力的涂层中有很大的应用前景[23, 24]。
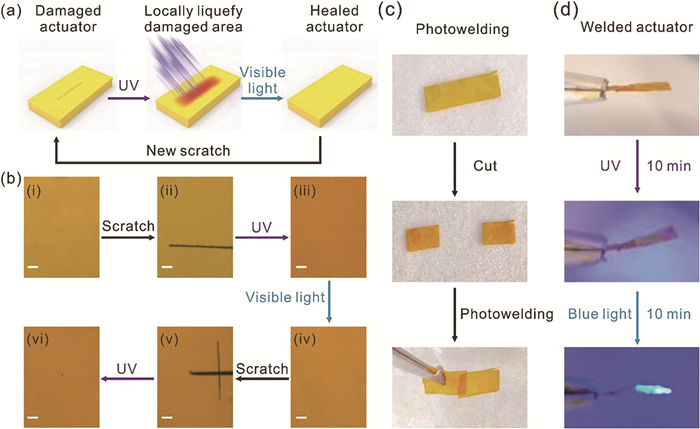
此项研究使用缠结的偶氮苯线性高分子制备了可修复以及可加工的光致动器,该致动器也表现出了光诱导可逆固液转变现象。与交联型的偶氮苯高分子相比,缠结的偶氮苯高分子表现出更好的机械性能。可以用光修复偶氮苯高分子致动器上的刮痕,并通过光图案化在致动器上制造微结构。该偶氮苯高分子致动器具有光致弯曲性能,还可引起微结构的可逆变形。高分子链缠结和光致可逆的固液转变过程相结合,为设计具有良好可再加工性和可修复性的致动器提供了新的思路。
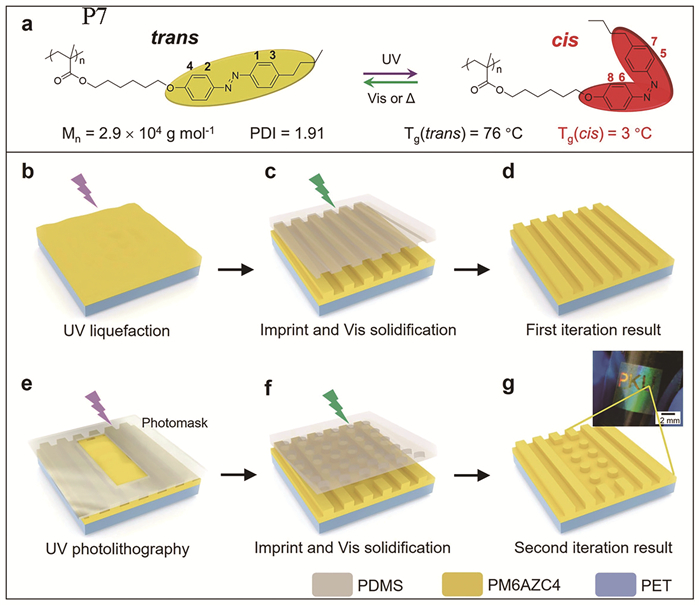
对于线性偶氮苯分子,Yang等[25]使用偶氮苯高分子P7实现了光控纳米压印(图 10)。他们将溶解在四氢呋喃(THF)中的P7旋涂在对苯二甲酸乙二醇酯(PET)基底上,使用紫外光照(图 10(b));然后将聚二甲基硅氧烷(PDMS)模具放在薄膜上,在外部应力下,液化的偶氮苯高分子流入模具的微阵列中(图 10(c));随后使用可见光固化P7,从而得到具有纳米压印的图案(图 10(d));其后,借助不同的光掩模经多次压印可实现不同的纳米图案(图 10(e~g))。
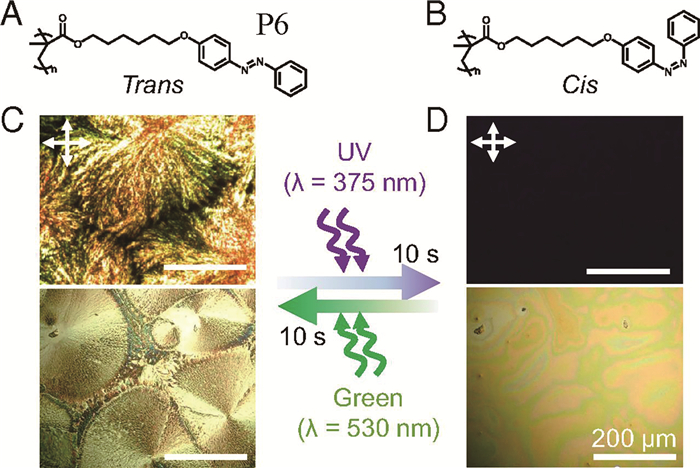
Shin等[26]发现,偶氮苯高分子P6的构型转变会导致该分子的热导率发生变化。P6经过紫外光照射后变为熔融态,绿光则使顺式构型P6分子异构化为反式构型,偶氮苯高分子则返回到结晶态(图 11(c))。进一步研究表明,平面结构的反式偶氮苯容易发生π-π堆积,而顺式偶氮苯则使得堆积减弱。顺式偶氮苯构型因苯环的旋转而破坏了偶氮苯基团间的π-π相互作用,从而形成热导率为0.1W·m-1·K-1的各向同性液体;绿光诱导形成的反式构型P6分子由相互交叉排列的平面偶氮苯侧链的晶体组成,因而具有3.5W·m-1·K-1的热导率。该研究揭示了偶氮苯高分子从晶体转变到熔融态热导率发生的变化,显示了其在热导开关方面的应用潜力。
偶氮苯基元的光致异构化能够调控高分子的Tg,从而在室温条件下可逆的调控高分子固液转变。这种光致固液转变现象具有广泛的应用,如制备自修复材料、转移印刷、光致热导开关器件、非热纳米压印、粘性材料以及光致动器等。
近年来的研究趋势表明,今后关于光化学在调控高分子性能方面的研究将越来越多,而具有光响应固液转变性能的偶氮苯高分子材料将是该领域的一个重要研究方向。
Hartley G. Nature, 1937, 140(3537):281.
Sun S, Liang S, Xu W, et al. Polym. Chem., 2019, 10(32):4389~4401. doi: 10.1039/C9PY00793H
Izumi A, Nomura R, Masuda T. Macromolecules, 2001, 34(13):4342~4347. doi: 10.1021/ma002101n
Schmidt H W. Angew. Chem. Int. Ed., 1989, 28(7):940~946. doi: 10.1002/anie.198909401
Ikeda T, Tsutsumi O. Science, 1995, 268(5219):1873~1875. doi: 10.1126/science.268.5219.1873
Akiyama H, Yoshida M. Adv. Mater., 2012, 24(17):2353~2356. doi: 10.1002/adma.201104880
Zhou H, Xue C, Weis P, et al. Nat. Chem., 2017, 9(2):145~151. doi: 10.1038/nchem.2625
Chen M, Yao B, Kappl M, et al. Adv. Funct. Mater., 2020, 30(4):1906752. doi: 10.1002/adfm.201906752
Zhou Y, Chen M S, Ban Q F, et al. ACS Macro Lett., 2019,8(8):968~972. doi: 10.1021/acsmacrolett.9b00459
Weis P, Hess A, Kircher G, et al. Chem. Eur. J., 2019, 25(46):10946~10953. doi: 10.1002/chem.201902273
Tian Y, Watanabe K, Kong X, et al. Macromolecules, 2002, 35(9):3739~3747. doi: 10.1021/ma011859j
Zhao Y, Qi B, Tong X, et al. Macromolecules, 2008, 41(11):3823~3831. doi: 10.1021/ma8000302
Ravi P, Sin S L, Gan L H, et al. Polymer, 2005, 46(1):137~146.
Ito S, Yamashita A, Akiyama H, et al. Macromolecules, 2018, 51(9):3243~3253. doi: 10.1021/acs.macromol.8b00156
Ito S, Akiyama H, Mori M, et al. Chem. Phys., 2019, 220(12):1900105.
Bandara H M D, Burdette S C. Chem. Soc. Rev., 2012, 41(5):1809~1825. doi: 10.1039/C1CS15179G
Akiyama H, Fukata T, Yamashita A, et al. J. Adhes., 2017, 93(10):823~830. doi: 10.1080/00218464.2016.1219255
Weis P, Hess A, Kircher G, et al. Chem. Eur. J., 2019, 25(46):10946~10953. doi: 10.1002/chem.201902273
Ito S, Akiyama H, Sekizawa R, et al. ACS Appl. Mater. Interf., 2018, 10(38):32649~32658. doi: 10.1021/acsami.8b09319
S Ito, H Akiyama, R Sekizawa et al. Polym. Chem., 2019, 57(7):806~813. doi: 10.1002/pola.29331
Wool R P. Macromolecules, 1993, 26(7):1564~1569. doi: 10.1021/ma00059a012
Xu W, Sun S, Wu S. Angew. Chem. Int. Ed., 2019, 58(29):9712~9740. doi: 10.1002/anie.201814441
Cui J, Drotlef D M, Larraza I, et al. Adv. Mater., 2012, 24(34):4601~4604. doi: 10.1002/adma.201200895
(a) Wu Z L, Buguin A, Yang H, et al. Adv. Funct. Mater., 2013, 23(24):3070~3076; (b) Kizilkan E, Strueben J, Staubitz A, et al. Sci. Robot., 2017, 2(2):eaak9454; (c) Liu D, Broer D J. Angew. Chem. Int. Ed., 2014, 53(18):4542~4546; (d) Yang R, Zhao Y. Angew. Chem. Int. Ed., 2017, 56(45):14202~14206.
Yang B, Cai F, Huang S, et al. Angew. Chem. Int. Ed., 2020, 59(10):4035~4042. doi: 10.1002/anie.201914201
Shin J, Sung J, Kang M, et al. PNAS, 2019, 116(13):5973~5978. doi: 10.1073/pnas.1817082116


图 2 (a) P1分子的可逆光致异构化,反式构型Tg高,顺式构型Tg低;(b)旋涂P1膜的紫外-可见光谱:光照前、紫外光照后(365nm, 67mW·cm-2, 1min)和可见光照后(530nm, 0.2mW·cm-2, 1min);(c)吸收强度随着紫外光与可见光交替照射的变化;(d) P1粉末在紫外光(365nm, 67mW·cm-2, 1min)与可见光(530nm, 0.2mW·cm-2, 1min)照射下的光学显微镜图片;(e) P1粉末在紫外光(365nm, 67mW·cm-2, 1min)照射后的共聚焦显微镜图片[7]
Figure 2 (a) Reversible photoisomerization of P1. P1is a solid with a high Tg in the trans state and is a liquid with a low Tg in the cis state; (b) UV-Vis absorption spectra of a spin-coated film of P1 before irradiation, after ultraviolet irradiation (365 nm, 67 mW·cm-2, 1 min) and subsequent visible light irradiation (530 nm, 0.2 mW·cm-2, 1 min); (c)Absorption changes under alternating ultraviolet and visible light irradiation; (d) Optical microscopy images of P1 powders under alternating ultraviolet (365 nm, 67 mW·cm-2) and visible light (530 nm, 5 mW·cm-2) irradiation, scale bars, 20μm; (e) Confocal microscope images of P1 powders before and after ultraviolet irradiation (365 nm, 67 mW·cm-2), ultraviolet irradiation induced the flow of P1 powders[7]

图 3 (a) 测量针尖与基底接触示意图;P1膜在照射前(b)、365nm光照射后(c)及530nm光照射后(d)的力-位置曲线[7]
Figure 3 (a) Schematic illustration of measuring force-piezo position curves; (b) Force-piezo position curves of the P1 film before irradiation; (c) after 365 nm ultraviolet irradiation (56 mW·cm-2, 6 min); (d) after 530 nm visible light irradiation (7 mW·cm-2, 6 min)[7]


图 5 (a) 偶氮苯高分子P1的光异构化导致可逆的固液转变(插图为紫外光照射前后P1粉末的光学显微镜图像);(b)光控粘合的示意图[9]
Figure 5 (a) Photoisomerization of the azopolymer P1 induces reversible solid-to-liquid transitions. The insets are optical microscopy images of P1 powders before and after UV irradiation; (b) Schematic illustration of the photoswitchable adhesion of P1-glued substrates[9]

图 6 (a) P1在两个石英基底上进行的剪切强度测试;(b)光控粘附性测试,紫外光照射左边,瓶子掉落;右边未经照射,瓶子未掉落;(c)粘合剂在溶剂和光辅助下重复使用[9]
Figure 6 (a) Lap joint shear strength tests with two quartz substrates glued with P1; (b) Snapshots show photocontrolled adhesion. Sample on the left: trans-P1-glued quartz substrates holding a bottle of water. The bottle fell after UV irradiation. Sample on the right: a control experiment showing that the P1-glued substrates held the bottle of water when P1 was not irradiated; (c) Light-assisted and solvent-assisted reuse of the adhesive[9]

图 7 (a) P1的化学结构以及光致异构化,光异构化使P1粉末从固体转变为液体,P1膜划痕的愈合以及无支撑膜的弯曲;(b)缠结示意图:低分子量的P1缠结较少,高分子量的P1链缠结较多;(c)从聚四氟乙烯的基底上剥离的偶氮苯高分子薄膜P1-10k和P1-100k;(d)P1-100k膜具有良好的透明度;(e)P1-100k具有良好的柔韧性;(f) P1-100k在90℃下可以拉伸[8]
Figure 7 (a) The chemical structure and photoisomerization of P1, Photoisomerization induced the solid to liquid transition of P1 powders, the healing of scratches on a P1 film, and the bending of a freestanding P1 film; (b) Schematic illustration: polymer chains of a low molecular weight P1 hardly entangle, while polymer chains of a high molecular weight P1 entangle; (c) Peeling off the azopolymer films P1-10k and P1-100k from Teflon substrates; (d) The freestanding film of P1-100k was highly transparent; (e) P1-100kis flexible; (f)P1-100kis stretchable at 90℃[8]


图 9 (a) 光诱导愈合示意图;(b)用光学显微镜观察P1-100k膜的修复(比例尺:20μm);(c)光焊接两片光致动器;(d)经紫外光(365nm,51mW·cm-2, 10min)和蓝光(470nm, 9mW·cm-2, 10min)照射后焊接的致动器发生弯曲[8]
Figure 9 (a) A schematic illustration of the photoinduced healing; (b) The photoinduced healing of scratches on a P1-100k film observed by optical microscopy. Scale bar: 20 μm; (c) The photowelding of an actuator that was cut into two pieces; (d) The photoinduced bending of the welded actuator under UV (365 nm, 51 mW·cm-2, 10 min) and blue light (470 nm, 9 mW·cm-2, 10 min) irradiation[8]

图 10 (a) 偶氮苯分子P7的化学结构以及光异构化;(b)紫外光液化;(c)压印以及可见光固化;(d)第一次压印结果;(e)使用光掩膜进行紫外光光刻;(f)压印以及可见光固化;(g)第二次压印结果[24]
Figure 10 (a) Chemical structure and photoisomerization of P7; (b) UV liquefaction; (c) imprinting, and visible-light solidification; (d) the result of the first imprint iteration; (e) UV photolithography with a photomask; (f) imprint and visible-light solidification; (g) the result of the second imprint iteration[24]

图 11 (a) 反式偶氮苯P6分子;(b)顺式偶氮苯P6分子;(c)反式偶氮苯P6分子,上:偏光显微镜示意图,下:光学显微镜示意图;(d)顺式偶氮苯P6分子,上:偏光显微镜示意图,下:光学显微镜示意图[25]
Figure 11 (a) Trans azobenzene P6; (b) Cis azobenzene P6; (c)Trans azobenzene P6, films under cross-polarized OM (Upper) and OM under continuous UV and green light illuminations (Lower); (d)Cis azobenzene P6, films under cross-polarized OM (Upper) and OM under continuous UV and green light illuminations (Lower)[25]

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

