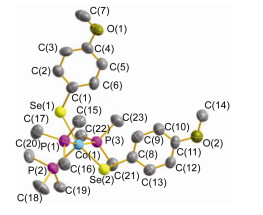
 图 1
配合物1的分子结构
Figure Figure1.
Molecular structure of complex 1
图 1
配合物1的分子结构
Figure Figure1.
Molecular structure of complex 1
引用本文:
栾化鑫, 孙宏建, 薛本静, 李晓燕. 三甲基膦支持的硒配位钴氢配合物的合成和性质[J]. 有机化学,
2017, 37(6): 1392-1397.
doi:
10.6023/cjoc201702047

Citation: Luan Huaxin, Sun Hongjian, Xue Benjing, Li Xiaoyan. Synthesis and Properties of a Se-Coordinated Hydrido Cobalt(Ⅲ) Complex Supported by Trimethylphosphine[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1392-1397. doi: 10.6023/cjoc201702047
Citation: Luan Huaxin, Sun Hongjian, Xue Benjing, Li Xiaoyan. Synthesis and Properties of a Se-Coordinated Hydrido Cobalt(Ⅲ) Complex Supported by Trimethylphosphine[J]. Chinese Journal of Organic Chemistry, 2017, 37(6): 1392-1397. doi: 10.6023/cjoc201702047
三甲基膦支持的硒配位钴氢配合物的合成和性质
摘要:
通过4-甲氧基苯硒酚和2-甲基苯硒酚分别与Co(PMe3)4反应生成了单核Co(Ⅱ)配合物[Co(4-MeOC6H4Se)2-(PMe3)3](1)和双核Co(Ⅰ)配合物[Co(PMe3)2(SeC6H4-2-Me)]2(2).配合物2与CO作用形成了配合物[Co(PMe3)2(CO)2(Se-C6H4-2-Me)](3).而苯硒酚与Co(PMe3)4反应制备了硒配位的钴氢配合物[mer-Co(H)(SePh)2(PMe3)3](4)和双核Co(Ⅰ)配合物[Co(PMe3)2(SePh)]2(5).初步研究表明4对于醛和酮的硅氢化反应有一定的催化效果.配合物1,3和4的分子结构通过X射线衍射进行了证实.
English
Synthesis and Properties of a Se-Coordinated Hydrido Cobalt(Ⅲ) Complex Supported by Trimethylphosphine
Abstract:
A mononuclear cobalt(Ⅱ) complex[Co(4-MeOC6H4Se)2(PMe3)3] (1) and a dinuclear cobalt diphenylselenolato complex[Co(PMe3)2(SeC6H4-2-Me)]2 (2) were obtained from the reaction of 4-methoxyselenophenol and 2-methylseleno-phenol with Co(PMe3)4, respectively. The dicarbonyl cobalt(Ⅰ) complex[Co(PMe3)2(CO)2(SeC6H4-2-Me)] (3) was formed through the reaction of complex 2 with carbon monoxide. A novel selenophenolato hydridocobalt(Ⅲ) complex[mer-Co(H)(SePh)2(PMe3)3] (4) and a dinuclear cobalt diphenylselenolato complex[Co(PMe3)2(SePh)]2 (5) were synthesized by the reaction of selenophenol with Co(PMe3)4. It was found that complex 4 could catalyze the hydrosilylation of aldehydes and ketones. The molecular structures of 1, 3 and 4 were determined by X-ray diffraction.
-
Key words:
- hydrido cobalt complex
- / selenophenol
- / Se-coordinated
- / hydrosilylation
- / carbonyl cobalt complex
-
过渡金属氢化物是一类重要的金属有机化合物, 被广泛应用于催化化学、合成化学和化学工业过程等各个领域.人们已经制备了一系列的过渡金属(尤其是贵金属)的氢化物, 并且研究了这些金属氢化物的催化作用[1~31].富电性金属铼的氢化物可以催化烯烃的加氢反应[8].傅尧等[9]在2015年利用Casey的铁氢催化剂实现了乙酰丙酸乙酯的转移氢化反应, 并最终得到γ-戊内酯.刘湘和李和兴等利用Cu(OAc)2•H2O为催化剂, 聚甲基氢硅氧烷(PMHS)为氢源, 在手性膦配体(R)-BINAP存在下实现了芳香酮的不对称硅氢化反应[10]. Milstein等[13]前不久报道了钌的[PNN]钳式氢化物可以有效地催化碱性水溶液中甲醇制氢的转化, 作为副产物的CO2可以利用碱液来吸收. Haenel, Kaska和Hall等[23]披露了铱的[PCP]钳式氢化物在均相催化条件下可以催化烷烃的脱氢反应.铑的氢化物在水溶液中既能与CO发生插入反应, 也能与醛和烯烃进行加成反应[30].相比较而言, 关于钴的金属氢化物的报道则比较少见[32~41]. Linehan等[37]在2014年发现了Co(dmpe)2H配合物在十分温和的条件下(101 kPa, 21 ℃)可以实现CO2的氢化, 产物为甲酸. Chirik等[38]发现钳式配体[CNC]的钴氢配合物可以催化烯烃的加氢反应.我们[39]曾经以硫代水杨醛为预配体合成了Co(Ⅲ)的氢化物, 首次研究了该配合物对于醛和酮中羰基的硅氢化反应的催化作用.我们还利用双C—H键的活化合成了[CNC]钳式Co(Ⅲ)氢配合物, 该配合物可以催化羰基化合物还原为醇的反应, 氢源为(EtO)3SiH[40].另外, 我们[41]通过Si—H键的活化合成了[PSiP]钳式Co(Ⅲ)的氢化物, 实验证明该氢化物可以在较温和条件下有效地催化Kumada偶联反应.
在早期的工作中, 我们[42]利用苯硫酚与CoCl-(PMe3)3或者与Co(PMe3)4的反应制得了硫原子配位的钴氢配合物, 并且研究了钴氢配合物的化学性质.硒和硫作为同一主族的元素, 尽管化学性质上具有一定的相似性, 作为不同周期的化学元素, 化学性质的差异性还是很明显的.为了拓展和积累我们在金属钴氢配合物领域里的研究工作, 本工作我们以苯硒酚为原料替代苯硫酚制备了新型钴氢配合物, 并且研究了相应氢化物的化学反应性质.
1 结果与讨论
1.1 Co(Ⅱ)配合物[Co(4-MeOC6H4Se)2(PMe3)3] (1) 的制备
将4-甲氧基苯硒酚在-78 ℃的正戊烷溶液中与[Co(PMe3)4]反应, 溶液的颜色立即由黄色转化为红棕色, 同时析出大量红棕色粉末.用乙醚萃取红棕色粉末, 过滤后在-20 ℃下由乙醚溶液中析出暗红色晶体, 为Co(Ⅱ)配合物1 (Eq. 1), 产率为51%.
配合物1的分子结构由单晶X射线衍射分析所证实(图 1).钴原子处于一个变形的四方锥(τ=0.08) 的四方面中心, P(2) 原子则占据该四方锥的顶点位置.由于键角P(1)—Co(1)—P(3) [167.10(9)°]和Se(1)—Co(1)—Se(2) [162.14(6)°]偏离了理想值(180°), 因此, 键角P(2)—Co(1)—P(3) [96.65(9)°], P(2)—Co(1)—P(1) [95.85(9)°], P(2)—Co(1)—Se(1) [98.12(7)°]和P(2)—Co(1)—Se(2) [99.68(7)°]也偏离了理想值(90°).四方面上四个配位键角(P(1)—Co(1)—Se(1) [88.63(7)°], P(1)—Co(1)—Se(2) [88.17(7)°], P(3)—Co(1)—Se(1) [86.49(7)°]和P(3)—Co(1)—Se(2) [92.83(7)°]之和为356.12°.这说明五个原子[Co(1) P(1) P(3) Se(1) Se(2)]的共平面性不好.两个4-MeOC6H4基团是向顶点配位键Co(1)—P(2) 的反方向伸展的, 显然, 这样可以使基团之间的排斥力最小, 能量最低.
图 1
配合物1的分子结构
Figure Figure1.
Molecular structure of complex 1
我们推测配合物1的可能生成机理如Scheme 1所示.在解离一分子三甲基膦配体的前提下, 4-甲氧基苯硒酚在钴中心发生氧化加成反应生成Co(Ⅱ)氢化物中间体1A. 1A不稳定, 通过单分子还原消除转化为中间物1B, 同时释放出氢气. Co(Ⅰ)配合物1B与第二个分子的4-甲氧基苯硒酚发生氧化加成反应生成不稳定的Co(Ⅲ)氢化物中间体1C. 1C通过释放氢气的单分子还原消除转化为最终产物1.配合物1具有成单电子是顺磁性的.但是实验中没有分离到中间体1A~1C.
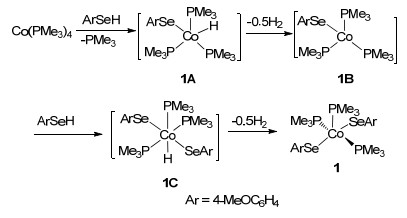 图式1
配合物1形成的可能机理
Scheme1.
Proposed mechanism of formation of complex 1
图式1
配合物1形成的可能机理
Scheme1.
Proposed mechanism of formation of complex 1
1.2 双核钴配合物[Co(PMe3)2(SeC6H4-2-Me)]2 (2) 的制备
相同反应条件下, 当选择2-甲基苯硒酚与[Co(PMe3)4]反应时, 既没有得到钴氢配合物, 也没有得到配合物1的类似物, 而是得到了双核Co(Ⅰ)配合物2 (Eq. 2), 产率为46%.当我们重新回顾我们早期的工作, 以硫酚为原料与[CoMe(PMe3)4]反应时分离得到的产物之一就是这类双核Co(Ⅰ)配合物[42]. Co(Ⅰ)配合物2是顺磁性物质, 其组成经过元素分析证实, 配合物2的结构应该与其硫配位的类似物相似[42].
为了进一步证实配合物2的结构, 我们设计了配合物2和CO的反应.在0.1 MPa的CO气压下, 配合物2在正戊烷的溶液中逐渐转化为双羰基配合物3 (Eq. 3).这一结果与我们早期的工作完全一致[42].配合物3为黄色晶体, 在空气中十分稳定.
在配合物3的红外光谱中, 两个端羰基强的红外吸收谱带分别处于1968和1893 cm-1, 该数值与其硫配位的类似物十分相近[42].单晶X射线衍射分析证实配合物3具有以钴为中心的三角双锥结构(图 2).两个三甲基磷配体处于轴向的位置.键角P(1)—Co(1)—P(2) 为175.31(2)°, 稍微偏离理想值180°.在赤道平面上, 由于苯环向C(1)—O(1) 键的方向伸展[Co(1)—Se(1)—C(3)=113.09(5)°], 使得键角Se(1)—Co(1)—C(1) [129.94(6)°]显著地大于键角Se(1)—Co(1)—C(2) [104.77(6)°].因此, 分子也失去了轴对称性.配合物3只有一个处于赤道平面的对称面.该反应也说明, 我们对于反应Eq. 2的推测是合理的.
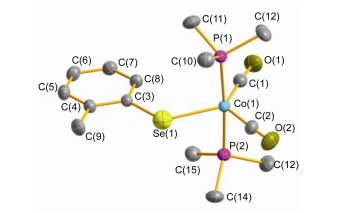 图 2
配合物3的分子结构
Figure Figure2.
Molecular structure of complex 3
图 2
配合物3的分子结构
Figure Figure2.
Molecular structure of complex 3
1.3 钴氢配合物[mer-Co(H)(SePh)2(PMe3)3] (4) 和双核钴配合物[Co(PMe3)2(PhSe)]2 (5) 的制备
将2 mol的苯硒酚与1 mol [Co(PMe3)4]配合物在-78 ℃的乙醚溶液中混合(Eq. 4), 温度逐渐升至室温, 并继续反应3 h, 得到棕黄色悬浊液, 过滤后在-20 ℃下由乙醚溶液中析出棕黄色晶体, 为Co(Ⅲ)的氢化物4, 产率为18%.母液抽干后继续用正戊烷萃取, 由滤液中析出配合物5, 产率40%.
在配合物4的红外光谱中, 化学键(Co—H)的伸缩振动出现在1955 cm-1处.与同系列的硫配位的钴氢配合物[ν(Co—H)=1977 cm-1]相比, 这种红移是由于硒取代配位所造成的[42].在配合物4的核磁共振氢谱中, 氢配体的质子共振信号以dt峰的形式出现在δ -9.90, 相应的偶合常数分别为2JPH=103和71 Hz.在配合物4的核磁共振磷谱中, 两组不同的共振磷信号以2:1的积分比例分别出现在δ -1.20和-7.55.这说明三个配位的PMe3配体具有两种不同的化学环境, 符合子午线排列(meridional)方式.
单晶的X射线衍射分析进一步证实配合物4的结构.配合物4具有六配位的变形八面体结构(图 3).三个配位的三甲基膦配体属于典型的子午线式排列.如果我们指认Se(2)—Co(1)—Se(1) [176.73(4)°]为轴向方向, 则中心钴原子则处于[HP(1) P(2) P(3)]四原子所确定的赤道平面中.键角Co(1)—Se(1)—C(1) 和Co(1)—Se(1)—C(7) 分别为114.6(2)°和113.3(2)°.由于氢配体的小体积使得两个苯环的伸展方向均为Co—H键的方向, 这样会使配合物4的结构最稳定, 能量最低.与键长Co(1)—P(1) [2.221(2) ]和Co(1)—P(3) [2.245(2) ]相比, 键长Co(1)—P(2) (2.270(2) )更长一点, 这可归因于负氢配体强的反位影响所带来的结果.我们早期的研究也证实了这个结论[42]. Co(Ⅲ)的氢化物4固体状态下在空气中相对稳定, 可以稳定存在大约2 h, 但在溶液中非常不稳定, 遇到空气立即分解.而且其溶液在室温下即使是氮气保护下也会逐渐分解, 这或许是其产率较低的原因.配合物5的组成经过元素分析表征初步证实, 5应该具有与2类似的结构.
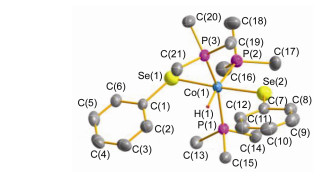 图 3
配合物4的分子结构
Figure Figure3.
Molecular structure of complex 4
图 3
配合物4的分子结构
Figure Figure3.
Molecular structure of complex 4
1.4 钴氢配合物4的催化性能探索
为了加深对钴氢配合物性质的认识, 我们初步探索了钴氢配合物4在醛和酮硅氢化反应中的催化性能.在5 mol% 4存在下, 以三乙氧基硅烷为氢源, 苯甲醛在50 ℃下反应4 h, 即可通过薄层色谱(TLC)检测到硅醚产物的存在.遗憾的是, 即使继续反应24 h仍未能转化完全.升高温度到60 ℃继续反应12 h仍没有明显变化.我们对底物进行了拓展, 结果见表 1.实验结果表明, 以5 mol% 4作为催化剂, 三乙氧基硅烷作为氢源, 在50 ℃的条件下, 钴氢配合物对醛的硅氢化反应有一定的催化效果, 但与我们之前报道的钴氢化合物的催化效果相比[39, 40]转化率较低, 而对酮的催化效果更差.我们推测催化效果不理想的原因是由钴氢配合物4的不稳定性造成的.
 表 1
配合物4对醛酮硅氢化反应的催化性质研究a
Table 1.
Catalytic property of complex 4 in hydrosilylation of aldehydes and ketones
表 1
配合物4对醛酮硅氢化反应的催化性质研究a
Table 1.
Catalytic property of complex 4 in hydrosilylation of aldehydes and ketones

推测配合物4催化醛酮硅氢化反应的可能机理如Scheme 2所示.首先, 一分子羰基化合物取代一分子三甲基膦配体生成π(C=O)配位的中间体a.然后, 氢配体亲核进攻羰基上的碳原子生成中间体b, 同时产生一个配位空缺.三乙氧基硅烷占据中间体b上的配位空穴形成中间体c.最后, 在三甲基膦存在下通过硅氢键活化消除一分子硅醚, 同时使催化剂4再生.
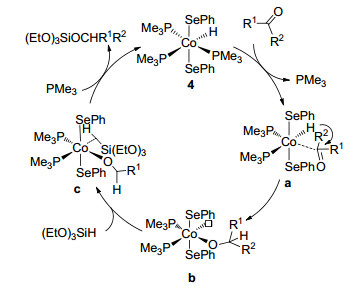 图式2
配合物4催化硅氢化反应的可能机理
Scheme2.
Proposed mechanism of hydrosilylation catalyzed by complex 4
图式2
配合物4催化硅氢化反应的可能机理
Scheme2.
Proposed mechanism of hydrosilylation catalyzed by complex 4
2 结论
利用苯硒酚和[Co(PMe3)4]的反应制得了Co(Ⅲ)氢配合物[mer-Co(H)(SePh)2(PMe3)3] (4). 4-甲氧基苯硒酚与[Co(PMe3)4]反应则生成了Co(Ⅱ)配合物[Co(4-MeO-C6H4Se)2(PMe3)3] (1).而2-甲基苯硒酚与[Co(PMe3)4]反应生成了双核钴配合物[Co(SeC6H4-2-Me)(PMe3)2]2 (2).初步探索了钴氢配合物4在醛和酮硅氢化反应中的催化性能.结果表明, 以5 mol%的4作为催化剂, 三乙氧基硅烷为氢源, 在50 ℃的条件下, 钴氢配合物对醛的硅氢化反应有一定的催化效果, 而对酮的催化效果较差.这或许是由钴氢配合物4的不稳定性造成的.双核钴配合物2和CO进一步反应, 生成双羰基钴配合物[Co(Se-C6H4-2-Me)(CO)2(PMe3)2] (3).上述实验结果说明, 各种苯硒酚的衍生物与[Co(PMe3)4]反应的产物是各具特色的.之所以会产生这种结果, 我们推测取代基在此起了很重要的作用. 4-甲氧基苯硒酚由于含有强给电子取代基致使钴氢中间体非常不稳定, 会迅速生成溶解度更差的配合物1. 2-甲基苯硒酚由于邻位含有给电子取代基致使空间位阻变大, 一方面阻碍了进一步结合邻甲基苯硒酚产生钴氢配合物, 再加上给电子基导致钴氢配合物稳定性变差, 从而使配合物2成为主要产物.到目前为止, 由于我们选择的苯硒酚种类有限, 还没有找到该反应的规律性.在该研究领域, 还有许多研究工作需要进一步地探索.
3 实验部分
3.1 仪器与试剂
核磁共振谱1H NMR, 31P NMR和13C NMR在Bruker AV 300 (300 MHz)核磁共振仪上测定; 元素分析则使用了Elementer Vario ELⅢ进行测量; 化合物的熔点用上海申光WRS-1C型熔点仪测定, 温度未经校正; 质谱则是在Agilent Technologies 6510 QTOF LC/MS型质谱仪中测出; 红外图谱用Bruker ALPHA FI-IR型测试仪测定.
所有溶剂均为分析纯级.乙醚、正戊烷、四氢呋喃使用前均在氮气氛中经钠-二苯甲酮回流处理.试剂邻溴甲苯、对溴苯甲醚从潍坊鸣冉化工有限公司购买, 溴苯购自国药集团化学试剂有限公司, 醛酮底物从上海海曲化工有限公司购买, 正十二烷购自天津科密欧化学试剂有限公司, 乙醚、四氢呋喃从天津富宇精细化工有限公司购买, 正戊烷购自天津市大茂化学试剂厂; PhSeH, o-MeC6H4SeH, p-MeOC6H4SeH和[Co(PMe3)4]根据文献[43, 44]方法合成.
除非特别说明, 所有试验均采用Schlenck实验技术, 在氮气保护的无水无氧条件下进行.实验中涉及到的CO为高纯气体.
3.2 实验方法
3.3 配合物晶体结构测定
单晶的X射线衍射实验是在STOE STADIVARI Cu或者Stoe IPDS2衍射仪上进行的.晶体结构和分子结构是利用Olex2[45]和ShelXS[46]程序包采用直接方法确定的.利用最小二乘法在ShelXL[47]程序包中进行了数据精修. CCDC-1518969 (1), 1511561 (3)和1511997 (4)的晶体学数据, 可以从下列地址中免费获取: CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44)1223-336-033; e-mail: deposit@ccdc.cam. ac.uk).
辅助材料(Supporting Information) 配合物的核磁共振氢谱、碳谱、磷谱以及红外谱图, 醇类产物的核磁共振氢谱、碳谱、质谱, 晶体结构的cif文件等.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
3.2.3 [Co(PMe3)2(CO)2(SeC6H4-2-Me)] (3)的合成
室温下, 将配合物2 (0.77 g, 1.00 mmol)溶于60 mL正戊烷中, 充入一个大气压的CO气体, 溶液颜色由红棕色变成黄色.继续搅拌反应36 h, 溶液经过滤后置于4 ℃.配合物3以黄色晶体的形式从正戊烷溶液中析出, 产量为0.59 g (67%). m.p.>125 ℃ (dec.); 1H NMR (300 MHz, C6D6) δ: 1.32 (s, 18H, PMe3), 2.94 (s, 3H, CH3), 7.13 (d, J=6.9 Hz, 3H, CH), 7.87 (d, J=7.2 Hz, 1H, CH); 31P NMR (121.5 MHz, C6D6) δ: 35.32; 13C NMR (75 MHz, C6D6, 298 K) δ: 18.71 (t, J=17.7 Hz, PMe3), 24.42 (s, Me), 122.63, 125.57, 128.94, 132.28, 138.63, 140.76, 202.03 (s, CO); IR (Nujol) ν: 1968, 1893 (CO), 1580 (C=C), 938 (PMe3) cm-1. Anal. calcd for C15H27CoO2P2Se: C 41.21, H 6.22; found C 41.47, H 5.89.
3.2.2 [Co(PMe3)2(SeC6H4-2-Me)]2 (2)的合成
将o-MeC6H4SeH (0.55 g, 3.2 mmol)溶于30 mL正戊烷溶液中, 在-78 ℃的条件下, 将其加入到30 mL Co(PMe3)4 (1.16 g, 3.2 mmol)的正戊烷溶液中.室温下搅拌反应3 h, 得到红棕色悬浊液.溶液经过滤后置于-20 ℃下, 配合物2以深红色晶体的形式在正戊烷溶液中析出, 产量0.56 g (46%). m.p.>74 ℃ (dec.); IR (Nujol) ν: 1584 (C=C), 944 (PMe3) cm-1. Anal. calcd for C26H50Co2P4Se2: C 40.96, H 6.61; found C 41.29, H 6.50.
3.2.7 催化醛酮硅氢化反应步骤
向含有3 mL THF的Schlenck管中依次加入1 mmol的醛或酮底物, 1.2 mmol的三乙氧基硅烷以及0.05 mmol的钴氢配合物4, 随后将上述体系置于50 ℃下加热搅拌, 并利用TLC进行检测.反应24 h后, 向反应体系中加入1 mL的MeOH以及5 mL的10%的NaOH水溶液.随后在60 ℃下, 将体系加热搅拌24 h进行水解.反应混合物用乙醚萃取后, 用无水Na2SO4干燥有机相, 利用气相色谱(GC)检测转化率, 并通过柱层析分离提纯催化产物.
3.2.4 [mer-Co(H)(SePh)2(PMe3)3] (4)和[Co(PMe3)2-(PhSe)]2 (5)的合成
将PhSeH (0.60 g, 3.8 mmol)溶于30 mL乙醚溶液中, 在-78 ℃的条件下, 将其加入到30 mL Co(PMe3)4 (0.69 g, 1.9 mmol)的乙醚溶液中.室温下搅拌反应3 h, 得到棕黄色悬浊液.溶液经过滤后置于-20 ℃.配合物4以棕黄色晶体的形式在乙醚溶液中析出, 产量为0.21 g (18%).母液在真空下转干溶剂后继续用30 mL正戊烷萃取, 过滤后置于-20 ℃.配合物5以深红色晶体的形式在正戊烷溶液中析出, 产量为0.28 g (40%).
4: m.p. 81~83 ℃ (dec.); 1H NMR (300 MHz, C6D6) δ: -9.90 (dt, 2JPtransH=103 Hz, 2JPcisH=71 Hz, 1H, Co-H), 1.22 (d, 2JPH=6.9 Hz, 9H, PMe3), 1.48 (t, |2JPH+4JPH|=3.6 Hz, 18H, PMe3), 7.11 (t, 3J=7.2 Hz, 2H, CH), 7.20 (t, 3J=7.8 Hz, 4H, CH), 8.44 (d, 3J=7.5 Hz, 4H, CH); 31P NMR (121.5 MHz, C6D6) δ: -7.55 (s, 1P, PMe3), -1.20 (s, 2P, PMe3); 13C NMR (75 MHz, C6D6) δ: 17.39 (d, 2J(PH)=21.8 Hz, PMe3), 20.93 (t, |2J(PH)+4J(PH)|=15.0 Hz, PMe3), 123.07, 127.93, 132.99, 137.24, 137.42; IR (Nujol) ν: 1955 (Co—H), 1571 (C=C), 938 (PMe3) cm-1. Anal. calcd for C21H38CoP3Se2: C 42.01, H 6.38; found C 41.67, H 6.63.
5: m.p.>71 ℃ (dec.); IR (Nujol) ν: 1572 (C=C), 934 (PMe3) cm-1. Anal. calcd for C24H46Co2P4Se2: C 39.26, H 6.31; found C 38.86, H 6.11.
3.2.1 [Co(4-MeOC6H4Se)2(PMe3)3] (1)的合成
将p-MeOC6H4SeH (0.57 g, 3.0 mmol)溶于30 mL正戊烷溶液中, 在-78 ℃的条件下, 将其加入到30 mL Co(PMe3)4 (0.55 g, 1.5 mmol)的正戊烷溶液中.反应迅速产生大量红棕色沉淀, 恢复室温后, 继续反应3 h.溶液经过滤后得到红棕色固体.将红棕色固体在乙醚溶液中于-20 ℃重结晶.配合物1以红棕色晶体的形式在乙醚溶液中析出, 产量0.51 g (51%). m.p.>200 ℃ (dec.); IR (Nujol) ν: 1586 (C=C), 944 (PMe3) cm-1. Anal. calcd for C23H41CoO2P3Se2: C 41.90, H 6.27; found C 41.52, H 6.09.
-
-
[1]
Bauer, I.; Knölker, H. J. Chem. Rev. 2015, 115, 3170. doi: 10.1021/cr500425u
-
[2]
Wang, W. H.; Himeda, Y.; Muckerman, J. T.; Manbeck, G. F.; Fujita, E. Chem. Rev. 2015, 115, 12936. doi: 10.1021/acs.chemrev.5b00197
-
[3]
Eberhardt, N. A.; Guan, H. R. Chem. Rev. 2016, 116, 8373. doi: 10.1021/acs.chemrev.6b00259
-
[4]
郭娜, 朱守非, 有机化学, 2015, 35, 1383. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344926.shtmlGuo, N.; Zhu, S. F. Chin. J. Org. Chem. 2015, 35, 1383 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344926.shtml
-
[5]
袁乾家, 张万斌, 有机化学, 2015, 36, 274. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345264.shtmlYuan, Q. J.; Zhang, W. B. Chin. J. Org. Chem. 2015, 36, 274 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345264.shtml
-
[6]
仉花, 孙宏建, 李晓燕, 有机化学, 2016, 36, 2843. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345675.shtmlZhang, H.; Sun, H. J.; Li, X. Y. Chin. J. Org. Chem. 2016, 36, 2843 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345675.shtml
-
[7]
Gunanathan, C.; Hölscher, M.; Pan, F.; Leitner, W. J. Am. Chem. Soc. 2012, 134, 14349. doi: 10.1021/ja307233p
-
[8]
Jiang, Y.; Huang, W.; Schmalle, H. W.; Blacque, O.; Fox, T.; Berke, H. Organometallics 2013, 32, 7043. doi: 10.1021/om400733u
-
[9]
Dai, N.; Shang, R.; Fu, M. C.; Fu, Y. Chin. J. Chem. 2015, 33, 405. doi: 10.1002/cjoc.v33.4
-
[10]
Liang, M. T.; Xia, X. F.; Liu, X.; Li, H. X. Chin. J. Chem. 2015, 33, 578. doi: 10.1002/cjoc.201500072
-
[11]
Yang, X. ACS Catal. 2012, 2, 964. doi: 10.1021/cs3000683
-
[12]
Huff, C. A.; Sanford, M. S. ACS Catal. 2013, 3, 2412. doi: 10.1021/cs400609u
-
[13]
Hu, P.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. ACS Catal. 2014, 4, 2649. doi: 10.1021/cs500937f
-
[14]
Arndtsen, B. A.; Bergman, R. G. Science 1995, 270, 1970. doi: 10.1126/science.270.5244.1970
-
[15]
Li, C.; Widjaja, E.; Garland, M. Organometallics 2004, 23, 4131. doi: 10.1021/om0342661
-
[16]
Filonenko, G. A.; Hensen, E. J.; Pidko, E. A. Catal. Sci. Technol. 2014, 4, 3474. doi: 10.1039/C4CY00568F
-
[17]
McGrady, G. S.; Guilera, G. Chem. Soc. Rev. 2003, 32, 383. doi: 10.1039/b207999m
-
[18]
Conifer, C.; Gunanathan, C.; Rinesch, T.; Hölscher, M.; Leitner, W. Eur. J. Inorg. Chem. 2015, 333.
-
[19]
Chen, H.; Schlecht, S.; Semple, T. C.; Hartwig, J. F. Science 2000, 287, 1995. doi: 10.1126/science.287.5460.1995
-
[20]
Hauwert, P.; Boerleider, R.; Warsink, S.; Weigand, J. J.; Elsevier, C. J. J. Am. Chem. Soc. 2010, 132, 16900. doi: 10.1021/ja1062407
-
[21]
Fan, L.; Parkin, S.; Ozerov, O. V. J. Am. Chem. Soc. 2005, 127, 16772. doi: 10.1021/ja0557637
-
[22]
Lersch, M.; Tilset, M. Chem. Rev. 2005, 105, 2471. doi: 10.1021/cr030710y
-
[23]
Haenel, M. W.; Oevers, S.; Angermund, K.; Kaska, W. C.; Fan, H. J.; Hall, M. B. Angew. Chem. 2001, 113, 3708. doi: 10.1002/(ISSN)1521-3757
-
[24]
Nielsen, M.; Alberico, E.; Baumann, W.; Drexler, H. J.; Junge, H.; Gladiali, S.; Beller, M. Nature 2013, 495, 85. doi: 10.1038/nature11891
-
[25]
Clarke, Z. E.; Maragh, P. T.; Dasgupta, T. P.; Gusev, D. G.; Lough, A. J.; Abdur-Rashid, K. Organometallics 2006, 25, 4113. doi: 10.1021/om060049z
-
[26]
Jiang, Y.; Blacque, O.; Fox, T.; Frech, C. M.; Berke, H. Organo-metallics 2009, 28, 5493. doi: 10.1021/om900458e
-
[27]
Guo, R.; Lough, A. J.; Morris, R. H.; Song, D. Organometallics 2004, 23, 5524. doi: 10.1021/om049460h
-
[28]
Burling, S.; Kociok-Köhn, G.; Mahon, M. F.; Whittlesey, M. K.; Williams, J. M. Organometallics 2005, 24, 5868. doi: 10.1021/om050600c
-
[29]
Jones, W. D. Inorg. Chem. 2005, 44, 4475. doi: 10.1021/ic050007n
-
[30]
Fu, X.; Wayland, B. B. J. Am. Chem. Soc. 2005, 127, 16460. doi: 10.1021/ja054548n
-
[31]
Li, X.; Chianese, A. R.; Vogel, T.; Crabtree, R. H. Org. Lett. 2005, 7, 5437. doi: 10.1021/ol052186i
-
[32]
Ding, K.; Brennessel, W. W.; Holland, P. L. J. Am. Chem. Soc. 2009, 131, 10804. doi: 10.1021/ja902812y
-
[33]
Du, J. Z.; Wang, L. B.; Xie, M. H.; Deng, L. Angew. Chem., Int. Ed. 2015, 54, 12640. doi: 10.1002/anie.201505937
-
[34]
Sun, J.; Luo, L.; Luo, Y.; Deng, L. Angew. Chem., Int. Ed. 2017, 56, 2720. doi: 10.1002/anie.v56.10
-
[35]
Scheuermann, M. L.; Semproni, S. P.; Pappas, I.; Chirik, P. J. Inorg. Chem. 2014, 53, 9463. doi: 10.1021/ic501901n
-
[36]
Tokmic, K.; Markus, C. R.; Zhu, L.; Fout, A. R. J. Am. Chem. Soc. 2016, 138, 11907. doi: 10.1021/jacs.6b07066
-
[37]
Jeletic, M. S.; Helm, M. L.; Hulley, E. B.; Mock, M. T.; Appel, A. M.; Linehan, J. C. ACS Catal. 2014, 4, 3755. doi: 10.1021/cs5009927
-
[38]
Yu, R. P.; Darmon, J. M.; Milsmann, C.; Margulieux, G. W.; Stieber, S. C. E.; DeBeer, S.; Chirik, P. J. J. Am. Chem. Soc. 2013, 135, 13168. doi: 10.1021/ja406608u
-
[39]
Niu, Q.; Sun, H.; Li, X.; Klein, H.-F.; Flörke, U. Organometallics 2013, 32, 5235. doi: 10.1021/om4005687
-
[40]
Zhou, H.; Sun, H.; Zhang, S.; Li, X. Organometallics 2015, 34, 1479. doi: 10.1021/om5011929
-
[41]
Xiong, Z.; Li, X.; Zhang, S.; Shi, Y.; Sun, H. Organometallics 2016, 35, 357. doi: 10.1021/acs.organomet.5b00937
-
[42]
Jiao, G.; Li, X.; Sun, H.; Xu, X. J. Organomet. Chem. 2007, 692, 4251. doi: 10.1016/j.jorganchem.2007.06.031
-
[43]
Foster, D. G. Org. Synth. 1944, 24, 89. doi: 10.15227/orgsyn.024.0089
-
[44]
Klein, H.-F.; Karsch, H. H. Chem. Ber. 1975, 108, 944. doi: 10.1002/(ISSN)1099-0682
-
[45]
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339. doi: 10.1107/S0021889808042726
-
[46]
Sheldrick, G. M. Acta Crystallogr. 2008, A64, 112.
-
[47]
Sheldrick, G. M. Acta Crystallogr. 2015, C71, 3.
-
[1]
-

图 1 配合物1的分子结构
Figure 1 Molecular structure of complex 1
Selected bond lengths (Å ) and bond angles (°): Co(1)—Se(1) 2.3907(14), Co(1)—Se(2) 2.3745(14), Co(1)—P(1) 2.224(2), Co(1)—P(2) 2.252(2), Co(1)—P(3) 2.233(2); P(1)—Co(1)—P(3) 167.11(7), Se(1)—Co(1)—Se(2) 162.15(6), P(2)—Co(1)—Se(1) 98.13(7), P(2)—Co(1)—Se(2) 99.67(7)

图 2 配合物3的分子结构
Figure 2 Molecular structure of complex 3
Selected bond lengths (Å ) and bond angles (°): Co—Se(1) 2.4384(3), Co(1)—P(1) 2.1909(5), Co(1)—P(2) 2.1933(5), Co(1)—C(1) 1.7417(17), Co(1)—C(2) 1.7603(17); P(1)—Co(1)—P(2) 175.312(18), C(1)— Co(1)—Se(1) 129.94(6), C(1)—Co(1)—C(2) 125.28(8), C(2)—Co(1)—Se(1) 104.77(6)

图 3 配合物4的分子结构
Figure 3 Molecular structure of complex 4
Selected bond lengths (Å ) and bond angles (°): Co(1)—H(1) 1.31(4), Co(1)—Se(1) 2.4015(8), Co(1)—Se(2) 2.4036(8), Co(1)—P(1) 2.2210(15), Co(1)—P(2) 2.2695(17), Co(1)—P(3) 2.2453(15); Se(1)—Co(1)—Se(2) 176.73(4), P(1)—Co(1)—Se(1) 95.80(4), P(1)—Co(1)—Se(2) 86.36(4), P(1)—Co(1)—P(2) 97.13(6), P(1)—Co(1)—P(3) 158.18(7), P(2)—Co(1)—Se(1) 88.15(5), P(2)—Co(1)—Se(2) 89.14(5), P(3)—Co(1)—Se(1) 85.00(4), P(3)—Co(1)—Se(2) 93.91(4), P(3)—Co(1)—P(2) 104.70(6)

图式2 配合物4催化硅氢化反应的可能机理
Scheme 2 Proposed mechanism of hydrosilylation catalyzed by complex 4
表 1 配合物4对醛酮硅氢化反应的催化性质研究a
Table 1. Catalytic property of complex 4 in hydrosilylation of aldehydes and ketones
 下载: 导出CSV
下载: 导出CSV
-






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 6
- 文章访问数: 1666
- HTML全文浏览量: 269
 下载:
下载:
