图式 1.
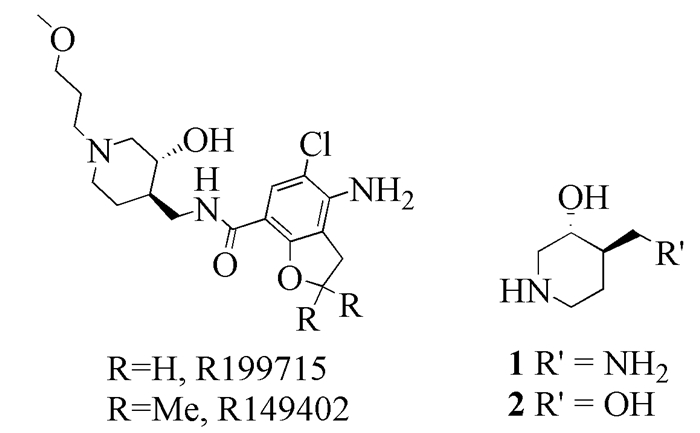
R149402、R199715及中间体1和2的结构
Scheme 1.
The structure of R149402, R199715, intermidiate 1 and 2

5-羟色胺(5-HT)受体,也被称为血清素受体,是一群于中枢神经系统中央处和末梢神经系统周边出现的G蛋白偶联受体及配体门控离子通道,广泛分布于人类外周器官,如胃肠道、血管和心脏等[1~3]。它们同时调节兴奋性和抑制性神经传导物质的传递。人体内存在多种5-HT受体亚型,目前已知的有7种,5-HT4是其中之一,位于胃肠道的外周,可介导胃肠道肌肉的收缩和舒张[4, 5]。
西沙必利(Cisapride)是5-HT4受体的激动剂,作为第三代全胃肠道促动力性药物,用于治疗食管及胃肠道各种运动障碍性疾病,副作用少,患者容易耐受。然而,自1989年上市后,因其存在引发致命性心律失常的风险而一度被撤市。因此,开发新型5-HT4受体激动剂,来替代西沙比利具有重要的意义[6~9]。De Maeyer等[10, 11]在进行药物化学研究过程中发现R149402和R199715具有很好的促进胃动力的的作用,并在对猪心脏变时性功能的研究中发现R149402和R199715没有表现出明显的正性肌力作用。Chai等[12]进一步发现,R199715表现出对人类心房5-HT4受体的拮抗作用,且不会引起正性肌力作用,能够消除引发心律失常的风险,有望用于非诱导正性肌力作用的胃肠运动障碍的治疗。目前R149402和R199715已作为治疗胃肠运动障碍的候选药物进入临床实验阶段。因此,R149402和R19975作为新型5-HT4受体激动剂,有望替代西沙必利,成为更为安全可靠的新一代胃肠道促动力性药物。因R149402和R199715都具有反式-4-胺甲基-哌啶-3-醇的结构,要构建这些结构就需要合成反-3, 4-哌啶二取代结构的中间体1和2。本文致力于关键中间体化合物2,即反-4-羟甲基哌啶-3-醇的合成和工艺优化。
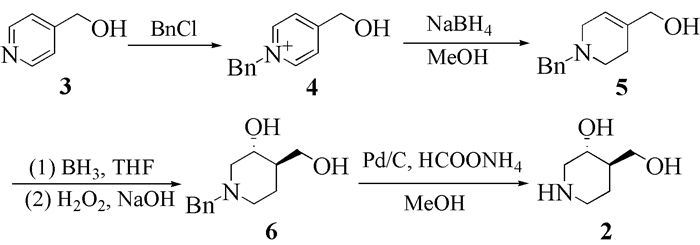
关于反-4-羟甲基哌啶-3-醇的合成报道极少。Gijsen等[13]报道了以4-羟甲基吡啶(3)为原料,与苄基氯在乙腈中反应生成吡啶鎓,再经硼氢化钠还原、硼氢化氧化、使用氢气脱苄基等反应来制备反-4-羟甲基哌啶-3-醇的方法。该方法总收率为42%,收率偏低,合成成本较高。本文在上述文献报道路线基础上,对苄基保护、硼氢化氧化、脱苄基反应进行了工艺优化,经四步反应合成了反-4-羟甲基哌啶-3-醇(2),合成路线如图式 2所示。该方法操作简便,收率高,成本低廉,有利于工业化生产。
Bruker Avance 400 MHz核磁共振谱仪(DMSO-d6或CDCl3为溶剂,TMS为内标);Agilent 1946 BESI-MS质谱仪;WRX-4显微熔点仪;Bruker Tensor 27傅立叶变换红外光谱仪。
4-羟甲基吡啶(含量>97%)、硼烷四氢呋喃络合物(1mol/L)、5%钯碳(含水量55%)购自萨恩化学技术(上海)有限公司;其余所用试剂均为市售分析纯级。
向50mL圆底烧瓶中加入5.00g(45.9mmol)4-羟甲基吡啶和7.00g(55.3mmol)苄基氯,反应体系在氩气保护下升温至70℃搅拌2h后,恢复至室温,加入乙醚洗涤除去过量的苄基氯,干燥后得到黄色油状液体9.17g,收率99.9%。1H NMR(360MHz,DMSO-d6)δ:4.81(s,2H),5.91(s,2H),7.35~7.43(m,3H),7.53~7.62(m,2H),8.06(d,J=6.7Hz,2H),9.20(d,J=6.7Hz,2H);IR(film)v/cm-1:3057,1640,1458,1074;LC-MS(ESI)m/z:201 [M+H]+。
用100mL甲醇将9.17g(45.85mmol)化合物4溶解,转移至500mL圆底烧瓶中,-20℃下搅拌30min,分批加入3.50g(92.11mmol)硼氢化钠,继续搅拌30min,恢复至室温,缓慢加入10mL水,减压蒸除有机溶剂后加100mL水溶解,二氯甲烷(3×100mL)萃取,有机相合并,用无水硫酸钠干燥,抽滤后蒸除有机溶剂,得到红色固体9.05g,收率97.2%,熔点63.5~64.4℃;1H NMR (360MHz,DMSO-d6)δ:2.01(s,2H),2.51(t,J=5.9Hz,2H),2.83~2.87(m,2H),3.53(s,2H),3.81(s,2H),4.67(t,1H),5.53(s,1H),7.21~7.25(m,1H),7.27~7.31(m,4H);IR(film)v/cm-1:3061,3027,2901,2800,1601,1494,1452,1050;LC-MS(ESI)m/z:204 [M+H]+。
在50mL三颈瓶中加入3.00g(14.77mmol)化合物5,用50mL四氢呋喃溶解,通入氩气保护,-30℃下搅拌30min后逐滴滴加30mL(30mmol)硼烷四氢呋喃,反应3h后转移至室温反应18h,冷却至-10℃后逐滴滴加8mL 55%氢氧化钠水溶液和8mL双氧水,搅拌30min后转移至60℃下加热回流3h,反应液转移至50mL圆底烧瓶中蒸除有机溶剂,加入50mL水溶解,乙酸乙酯(3×40mL)萃取,合并有机相,用无水硫酸钠干燥,过滤,蒸除有机溶剂,在乙酸乙酯/异丙醚(体积比1:50)中析晶,得白色针状结晶1.95g,收率为59.7%,熔点103.8~104.5℃;1H NMR(400MHz,CDCl3)δ:1.24(qd,J=13.0、3.7 Hz,1H),1.44~1.52(m,2H),1.81(t,J=10.3Hz,1H),1.92(td,J=11.7、2.8 Hz,1H),2.76(br d,J=11.4Hz,1H),2.81(br s,1H),2.93(ddd,J=10.7、4.4、1.5 Hz,1H),3.40(br s,1H),3.45(d,J=12.9 Hz,1H),3.51(d,J=12.9 Hz,1H),3.58~3.69(m,3H),7.16~7.27(m,5H);IR(film) v/cm-1:3062,3028,2914,2804,1600,1494,1449,1099,1038;LC-MS(ESI)m/z:222 [M+H]+。
在50mL圆底烧瓶中加入1.95g(8.82mmol)化合物6和1.10g(17.5mmol)甲酸铵,用甲醇20mL溶解,加入0.20g钯碳,通入氩气保护,60℃下加热回流2h后冷却至室温,硅藻土过滤,减压加热蒸除有机溶剂和体系中易挥发物质即可得黄色油状液体1.11g,收率为96.1%。1H NMR(400MHz,CDCl3)δ:1.16~1.29(m,2H),1.30~1.40(m,1H),2.30(dd,J=11.7、10.7 Hz,1H),2.41(t,J=11.8Hz,1H),2.63(td,J=12.6、3.0 Hz,1H),3.04(brd,J=12.0 Hz,1H),3.20(ddd,J=11.1、4.6、1.7 Hz,1H),3.59(dd,J=10.9、4.1 Hz,1H),3.73(dd,J=10.9、3.1 Hz,1H),3.82(td,J=9.6、4.6 Hz,1H);IR(film) v/cm-1:3275,2922,1105,1055,1028;LC-MS (ESI)m/z:132 [M+H]+。
苄基保护反应在70℃下进行。实验过程中发现当温度上升到3的熔点(52℃)附近时,3即可全溶于1.2倍摩尔当量的苄基氯中,在不额外使用有机溶剂的条件下继续反应,收率能够达到99.9%,而参考文献中使用乙腈作为溶剂溶解3和苄基氯后进行反应,收率为97%[13]。与文献方法相比,本方法不仅收率略有提高,而且制备成本低廉,后处理简单,产生三废少。
由5合成6的反应中,对硼氢化氧化后的水解步骤进行了优化。文献报道方法为依次加入3mol/L和50%的氢氧化钠水溶液,收率为50%[13]。本文采取一次性投料方法,考察了氢氧化钠用量对收率的影响。随着氢氧化钠用量的增加,反应收率不断增加,当加入的氢氧化钠溶液接近饱和浓度55%时,即可达到最高收率59.7%(表 1)。由此可见,硼氢化氧化后的水解过程中,体系内碱的用量增加有助于提高反应收率。
 下载:
导出CSV
下载:
导出CSV
| 氢氧化钠浓度/% | 用量/mL | 收率/% |
| 30 | 8 | 28.8 |
| 40 | 8 | 46.6 |
| 50 | 8 | 56.7 |
| 55 | 8 | 59.7 |
使用钯碳脱苄基的方法主要用到的供氢体有氢气和甲酸铵两种。本文考察了两种供氢体对反应收率的影响,结果表明,分别以氢气和甲酸铵为供氢体时,反应的产率分别为98%和96.1%。文献中使用的供氢体为氢气[13]。使用氢气进行脱苄基是非均相反应,需要在高于常压的密闭容器中进行,对反应设备有较苛刻的要求,伴随一定的危险性。相较而言,使用甲酸铵的脱苄基反应为常压下的均相反应,对反应设备无特殊要求。此外,甲酸铵为常规试剂,价格低廉,危险性小,有利于降低工艺成本。因此,本文在收率几乎相同的前提下,选择甲酸铵用于脱苄基反应。
以4-吡啶甲醇为起始原料,合成了5-HT4受体激动剂的关键中间体反-4-羟甲基哌啶-3-醇,总收率55.7%。经优化后的合成工艺具有成本低廉、操作简单、收率高等优点,为新型5-HT4受体激动剂药物的合成提供了良好的途径。
Y Sakurai-Yamashita, K Yamashita, M Yoshimura et al. Life Sci., 2000, 66(1):31~34.
K Y Chan, R De Vries, F P J Leijten et al. Eur. J. Pharm., 2009, 619(1~3):61~67.
T Bach, T Syversveen, A M Kvingedal et al. Naunyn-Schmiedeberg's Arch. Pharm., 2001, 363(2):146~160. doi: 10.1007/s002100000299
宋清, 张晓文, 张绒. 甘肃医药, 2010, 29(1):1~4. http://www.cnki.com.cn/Article/CJFDTotal-gsyy201001003.htm
陆伦根. 国际消化病杂志, 2000, 20(2):114~116. http://www.cnki.com.cn/Article/CJFDTotal-GWXH200002015.htm
R W Mc Callum. Am. J. Gastroenterol., 1991, 86(2):135~149.
赵香花, 张晓冬, 黄矛. 解放军药学学报, 2009, 25(5):391~395. http://www.cnki.com.cn/Article/CJFDTotal-JFJN200905007.htm
S Mohammad, Z Zhou, Q Gong. Am. J. Physiol. Heart C, 1997, 273(5):H2534~H2538.
E Farrington. Pediatric Nurs., 1996, 22(3):256~256.
J H De Maeyer, N H Prins, J A Schuurkes et al. Pharmacol. Exp. Ther., 2006, 317(3):955~964. doi: 10.1124/jpet.106.101329
J H De Maeyer, R Straetemans R, J A Schuurkes et al. Br. J. Pharmacol., 2006, 147(2):140~157. doi: 10.1038/sj.bjp.0706497
W Chai, K Y Chan, R De Vries et al. Life Sci., 2012, 90(13~14):538~544.
H J M Gijsen, M J A De Cleyn et al. Tetrahedron, 2008, 64(10):2456~2464. doi: 10.1016/j.tet.2007.12.055

图式 1 R149402、R199715及中间体1和2的结构
Scheme 1 The structure of R149402, R199715, intermidiate 1 and 2
表 1 碱的用量对化合物6收率的影响
Table 1. The effect of amount of base on the yield of 6
| 氢氧化钠浓度/% | 用量/mL | 收率/% |
| 30 | 8 | 28.8 |
| 40 | 8 | 46.6 |
| 50 | 8 | 56.7 |
| 55 | 8 | 59.7 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们